Yoon Hongsup, Gerdes Lisa Ann, Beigel Florian, Sun Yihui, Kövilein Janine, Wang Jiancheng, Kuhlmann Tanja, Flierl-Hecht Andrea, Haller Dirk, Hohlfeld Reinhard, Baranzini Sergio E, Wekerle Hartmut, Peters Anneli
Institute of Clinical Neuroimmunology, University Hospital Ludwig-Maximilians-Universität München, Martinsried 82152, Germany.
Biomedical Center, Faculty of Medicine, Ludwig-Maximilians-Universität München, Martinsried 82152, Germany.
Proc Natl Acad Sci U S A. 2025 May 6;122(18):e2419689122. doi: 10.1073/pnas.2419689122. Epub 2025 Apr 21.
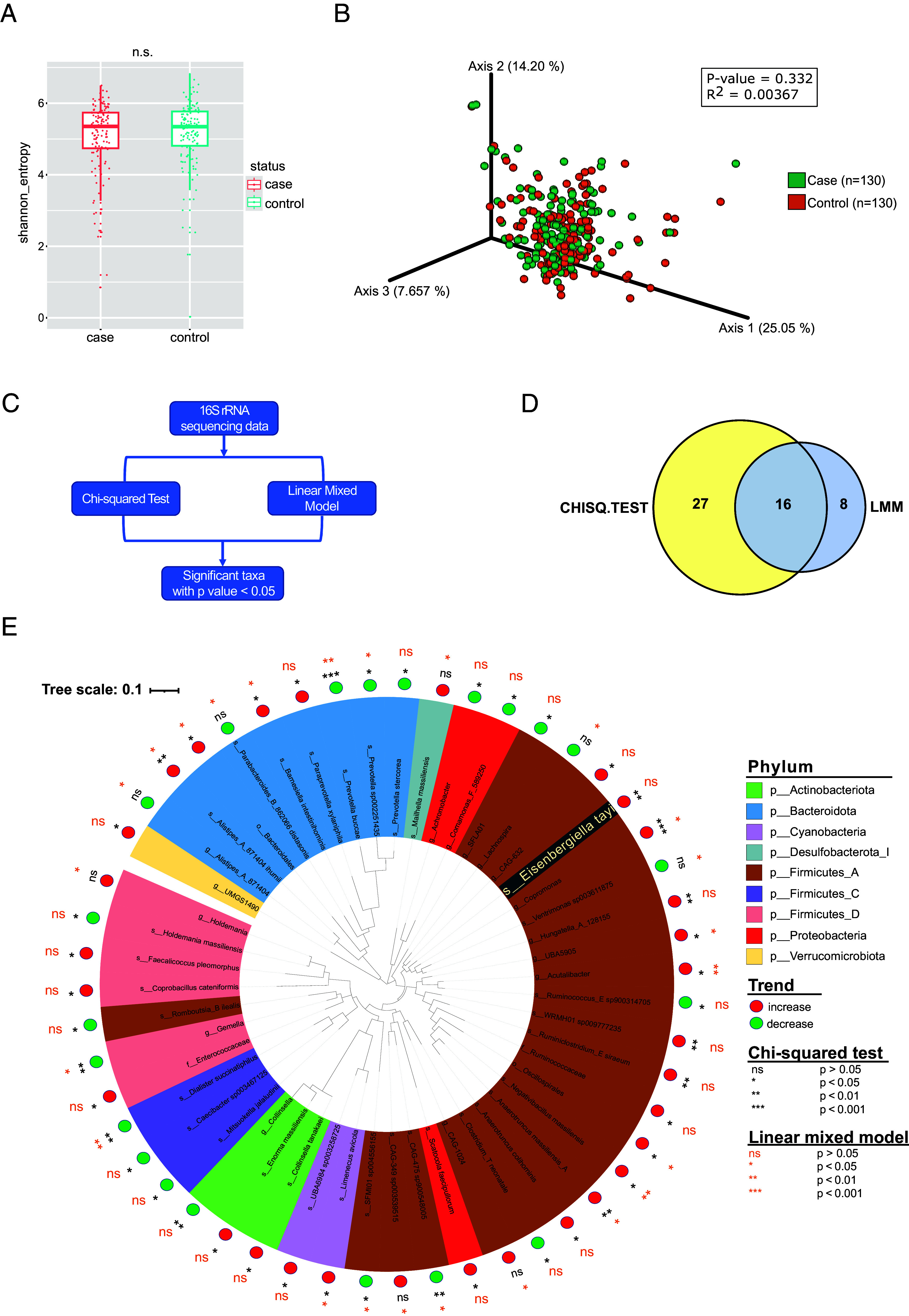
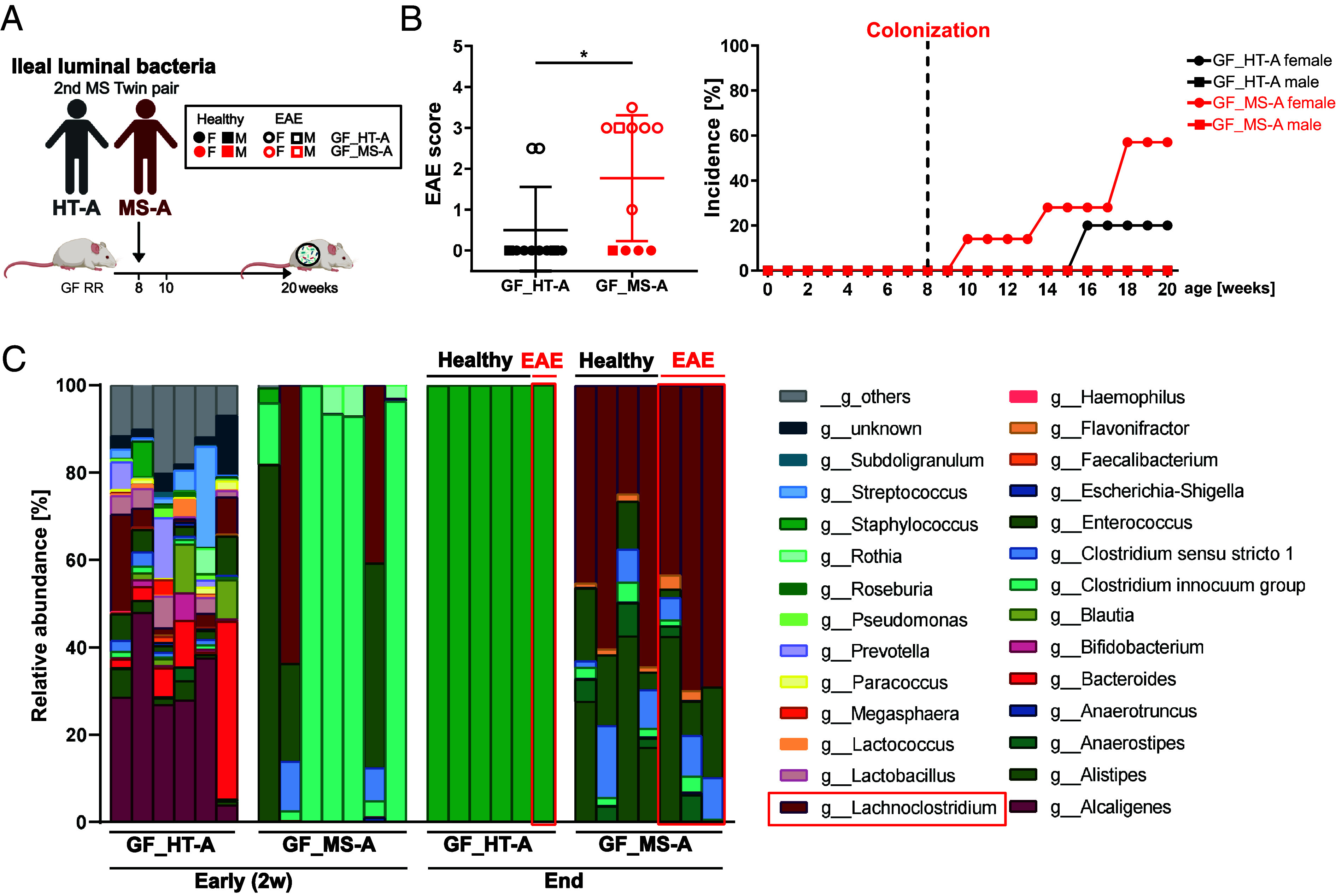
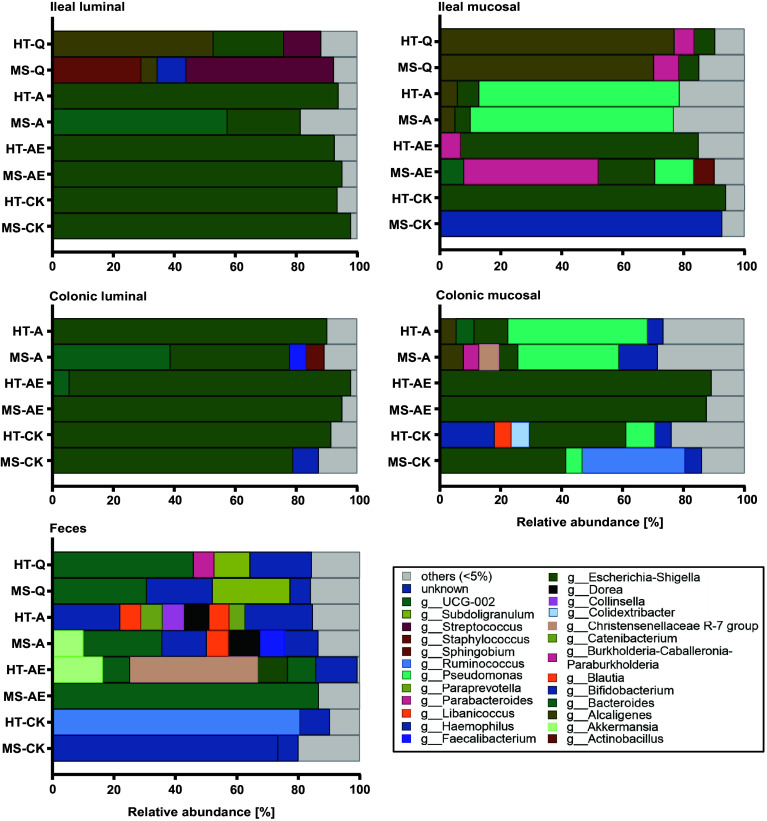
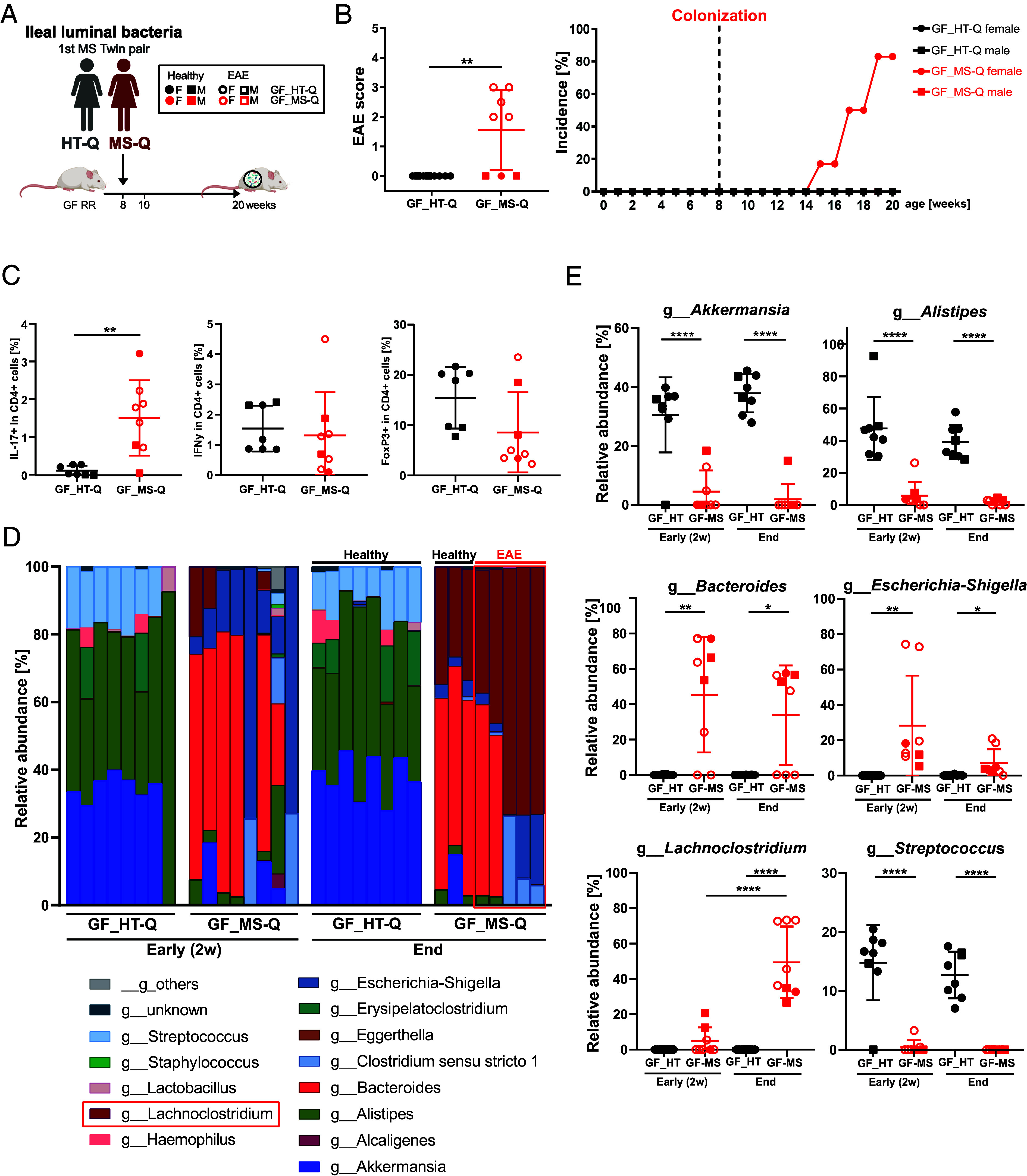
We developed a two-tiered strategy aiming to identify gut bacteria functionally linked to the development of multiple sclerosis (MS). First, we compared gut microbial profiles in a cohort of 81 monozygotic twins discordant for MS. This approach allowed to minimize confounding effects by genetic and early environmental factors and identified over 50 differently abundant taxa with the majority of increased taxa within the . These included taxa previously described to be associated with MS ( and ), along with newly identified taxa, such as and . Second, we interrogated the intestinal habitat and functional impact of individual taxa on the development of MS-like disease. In an exploratory approach, we enteroscopically sampled microbiota from different gut segments of selected twin pairs and compared their compositional profiles. To assess their functional potential, samples were orally transferred into germfree transgenic mice prone to develop spontaneous MS-like experimental autoimmune encephalomyelitis (EAE) upon bacterial colonization. We found that MS-derived ileal microbiota induced EAE at substantially higher rates than analogous material from healthy twin donors. Furthermore, female mice were more susceptible to disease development than males. The likely active organisms were identified as and members of the Lachnospiraceae family. Our results identify potentially disease-facilitating bacteria sampled from the ileum of MS affected twins. The experimental strategy may pave the way to functionally understand the role of gut microbiota in initiation of MS.
我们制定了一项分两个层次的策略,旨在识别与多发性硬化症(MS)发展在功能上相关的肠道细菌。首先,我们比较了81对患MS的单卵双胞胎的肠道微生物谱。这种方法能够将遗传和早期环境因素造成的混杂效应降至最低,并识别出50多种丰度不同的分类群,其中大多数丰度增加的分类群在……范围内。这些分类群包括先前描述的与MS相关的分类群(……和……),以及新识别出的分类群,如……和……。其次,我们探究了单个分类群的肠道栖息地及其对类MS疾病发展的功能影响。在一种探索性方法中,我们通过内镜从选定双胞胎对的不同肠道段采集微生物群,并比较它们的组成谱。为了评估它们的功能潜力,将样本口服转移到无菌转基因小鼠体内,这些小鼠在细菌定植后容易自发发展为类MS实验性自身免疫性脑脊髓炎(EAE)。我们发现,来自MS患者回肠的微生物群诱发EAE的比率明显高于来自健康双胞胎供体的类似物质。此外,雌性小鼠比雄性小鼠更容易发病。可能起作用的微生物被确定为毛螺菌科的……和……成员。我们的结果确定了从患MS的双胞胎回肠中采样的可能促进疾病的细菌。该实验策略可能为从功能上理解肠道微生物群在MS发病中的作用铺平道路。