Teschner David, Gomez-Zepeda David, Łącki Mateusz K, Kemmer Thomas, Busch Anne, Tenzer Stefan, Hildebrandt Andreas
Institute of Computer Science, Johannes-Gutenberg University, 55128 Mainz, Germany.
Institute for Quantitative and Computer Biosciences (IQCB), Johannes-Gutenberg University, 55128 Mainz, Germany.
J Proteome Res. 2025 May 2;24(5):2358-2368. doi: 10.1021/acs.jproteome.4c00966. Epub 2025 Apr 22.
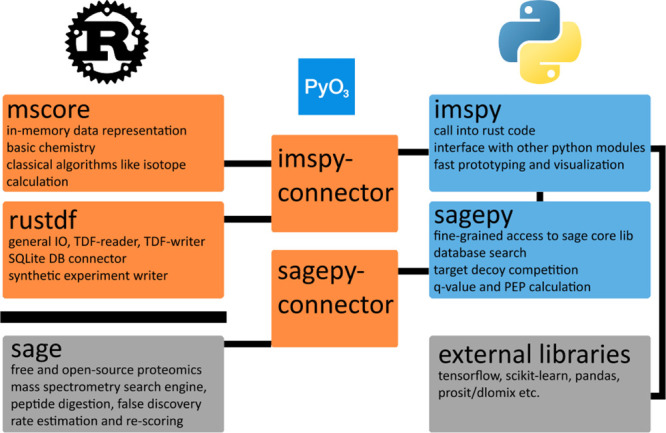
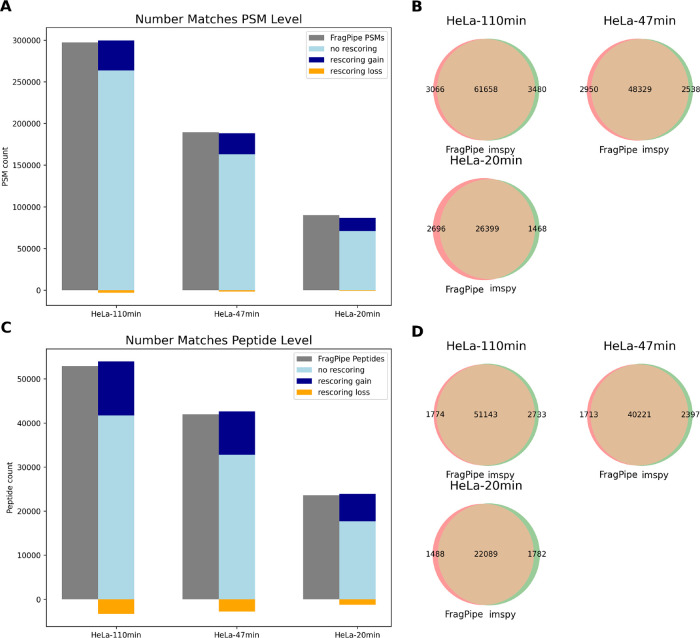
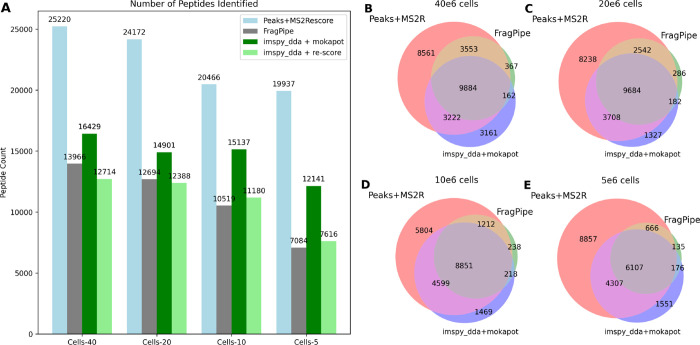
Mass spectrometry is essential for analyzing and quantifying biological samples. The timsTOF platform is a prominent commercial tool for this purpose, particularly in bottom-up acquisition scenarios. The additional ion mobility dimension requires more complex data processing, yet most current software solutions for timsTOF raw data are proprietary or closed-source, limiting integration into custom workflows. We introduce rustims, a framework implementing a flexible toolbox designed for processing timsTOF raw data, currently focusing on data-dependent acquisition (DDA-PASEF). The framework employs a dual-language approach, combining efficient, multithreaded Rust code with an easy-to-use Python interface. This allows for implementations that are fast, intuitive, and easy to integrate. With imspy as its main Python scripting interface and sagepy for Sage search engine bindings, rustims enables fast, integrable, and intuitive processing. We demonstrate its capabilities with a pipeline for DDA-PASEF data including rescoring and integration of third-party tools like the Prosit intensity predictor and an extended ion mobility model. This pipeline supports tryptic proteomics and nontryptic immunopeptidomics data, with benchmark comparisons to FragPipe and PEAKS. Rustims is available on GitHub under the MIT license, with installation packages for multiple platforms on PyPi and all analysis scripts accessible via Zenodo.
质谱分析对于生物样品的分析和定量至关重要。timsTOF平台是用于此目的的一种重要商业工具,特别是在自下而上的采集场景中。额外的离子淌度维度需要更复杂的数据处理,但目前大多数用于timsTOF原始数据的软件解决方案都是专有的或封闭源代码的,这限制了其集成到自定义工作流程中。我们引入了rustims,这是一个实现灵活工具箱的框架,旨在处理timsTOF原始数据,目前专注于数据依赖采集(DDA-PASEF)。该框架采用双语方法,将高效的多线程Rust代码与易于使用的Python接口相结合。这使得实现快速、直观且易于集成。以imspy作为其主要的Python脚本接口,并使用sagepy进行Sage搜索引擎绑定,rustims实现了快速、可集成且直观的数据处理。我们通过一个用于DDA-PASEF数据的流程展示了它的功能,该流程包括重新评分以及整合第三方工具(如Prosit强度预测器和扩展离子淌度模型)。此流程支持胰蛋白酶蛋白质组学和非胰蛋白酶免疫肽组学数据,并与FragPipe和PEAKS进行了基准比较。rustims在GitHub上根据MIT许可提供,在PyPi上有适用于多个平台的安装包,并且所有分析脚本都可通过Zenodo获取。