Zheng Liping, Yan Yifan, Li Qun, Du Junyang, Lu Xiaosong, Xu Li, Xie Qunhui, Chen Yangsheng, Zhang Aiguo, Zhao Bin
State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China.
University of Chinese Academy of Sciences, Beijing 100049, China.
Microorganisms. 2025 Apr 10;13(4):869. doi: 10.3390/microorganisms13040869.
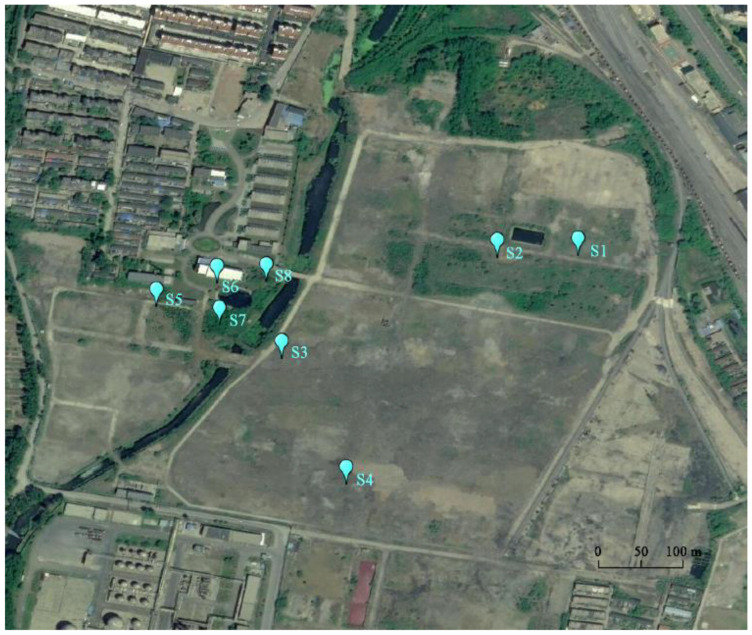
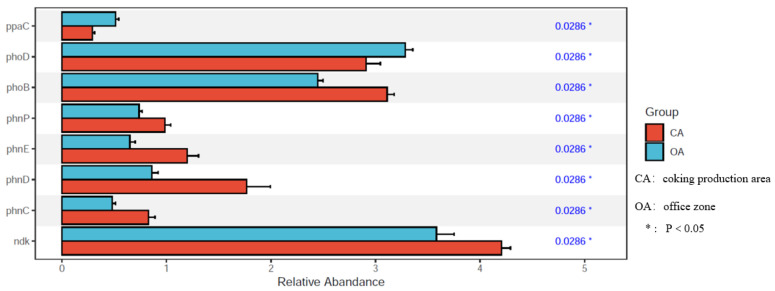
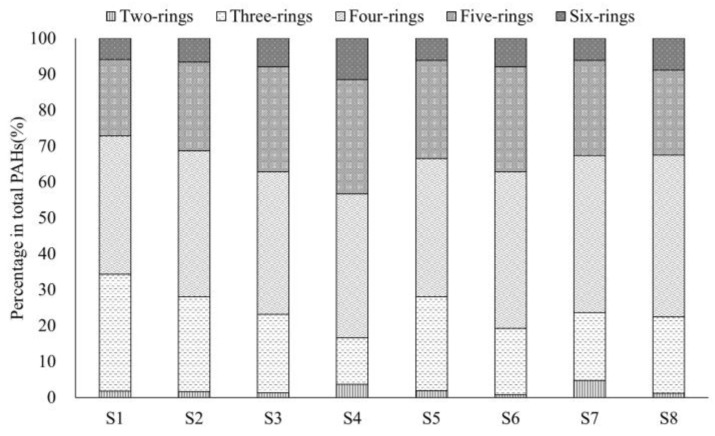
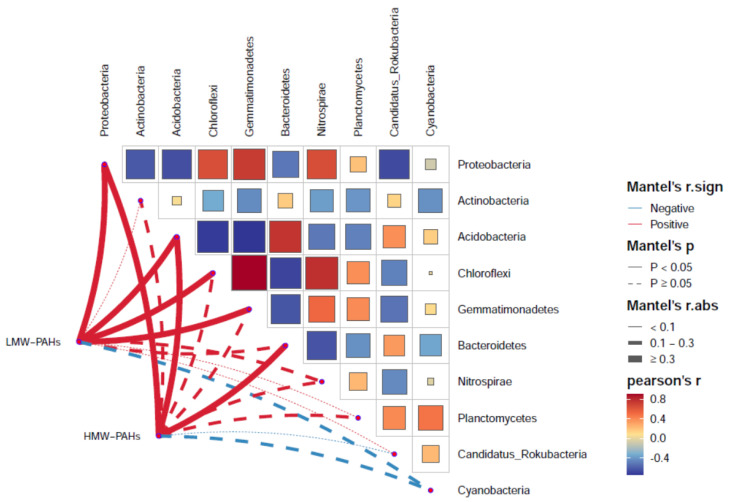
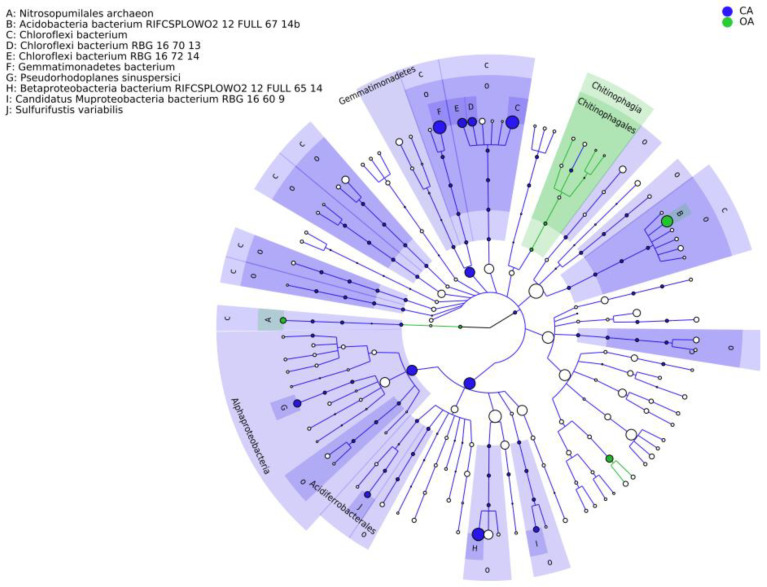
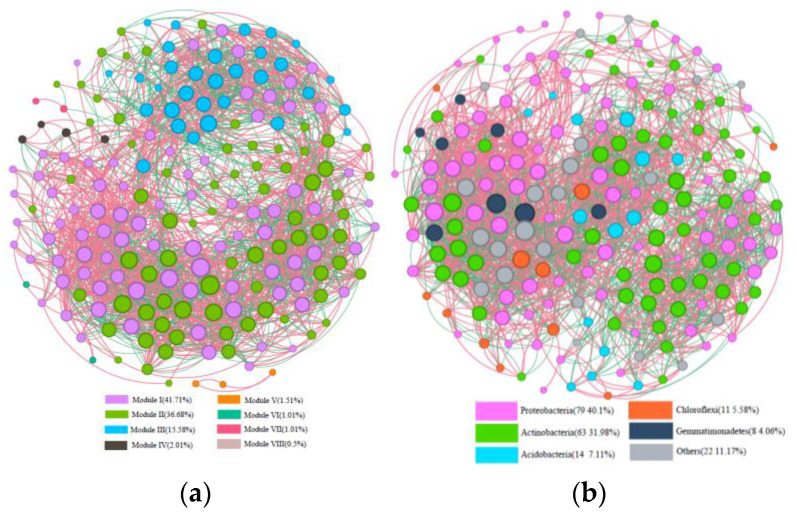
PAH contamination from coking plants have received widespread attention. However, the microbial diversity, co-occurrence patterns, and functional genes of bacteria in aged coking contaminated soils by PAHs are still not clear. In our study, we used a macro-genetic approach to detect PAH-contaminated soils from both a coking production area (CA group) and an office zone (OA group) in an abandoned coking plant, and analyzed the characteristic bacteria and function genes, microbial network interaction patterns, and soil P-cycling in long-term PAH-contaminated soils. The results revealed that were significantly positively correlated with PAHs and rifcsplowo2 12 full 6514, candidatus RBG16609, and , which belong to , were characteristic bacteria in PAH-contaminated soils. The phn, which is the PAH degradation gene, was abundantly expressed in the PAH-contaminated soil. The gene cluster genes (, , and ) were significantly expressed in the CA group of PAH-contaminated soils ( < 0.05). By integrating microbial diversity, network structure, and functional genes, it offers a comprehensive understanding of soil ecosystem response indicators to prolonged PAH stress. The results of this study will provide new ideas for constructing an assessment index system for soil health and screening biomarkers for PAH-contaminated soils.
焦化厂的多环芳烃(PAH)污染已受到广泛关注。然而,长期受PAHs污染的焦化土壤中细菌的微生物多样性、共生模式和功能基因仍不清楚。在我们的研究中,我们采用宏基因组方法检测了一个废弃焦化厂的焦化生产区(CA组)和办公区(OA组)的PAH污染土壤,并分析了长期受PAH污染土壤中的特征细菌和功能基因、微生物网络相互作用模式以及土壤磷循环。结果表明,[此处原文缺失具体内容]与PAHs显著正相关,[此处原文缺失具体内容]以及属于[此处原文缺失具体内容]的candidatus RBG16609和[此处原文缺失具体内容]是PAH污染土壤中的特征细菌。作为PAH降解基因的phn在PAH污染土壤中大量表达。基因簇基因([此处原文缺失具体内容]、[此处原文缺失具体内容]和[此处原文缺失具体内容])在PAH污染土壤的CA组中显著表达(P<0.05)。通过整合微生物多样性、网络结构和功能基因,它提供了对土壤生态系统对长期PAH胁迫响应指标的全面理解。本研究结果将为构建土壤健康评估指标体系和筛选PAH污染土壤的生物标志物提供新思路。