Srinivasa Vatsala Rangachar, Griffith Marissa P, Sundermann Alexander J, Mills Emma, Raabe Nathan J, Waggle Kady D, Shutt Kathleen A, Phan Tung, Wang-Erickson Anna F, Snyder Graham M, Van Tyne Daria, Pless Lora Lee, Harrison Lee H
Microbial Genomic Epidemiology Laboratory, Center for Genomic Epidemiology, University of Pittsburgh, Pittsburgh, PA, USA.
Division of Infectious Diseases, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA.
medRxiv. 2025 Apr 25:2025.04.20.25325828. doi: 10.1101/2025.04.20.25325828.
Respiratory virus transmission in healthcare settings is not well understood. To investigate the transmission dynamics of common healthcare-associated respiratory virus infections, we performed retrospective whole genome sequencing (WGS) surveillance at one pediatric and two adult teaching hospitals in Pittsburgh, PA.
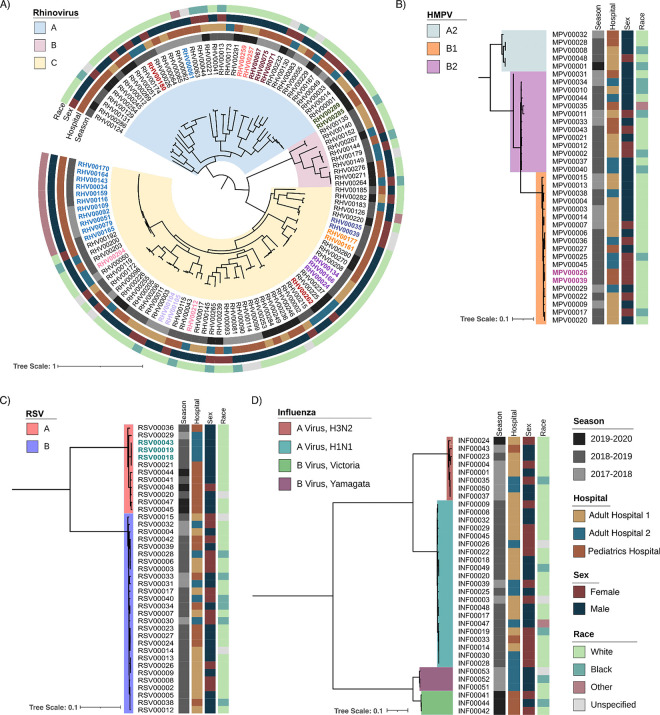
From January 2, 2018, to January 4, 2020, nasal swab specimens positive for rhinovirus, influenza, human metapneumovirus (HMPV), or respiratory syncytial virus (RSV) from patients hospitalized for ≥3 days were sequenced on Illumina platform. High-quality genomes were assessed for genetic relatedness using ≤3 single nucleotide polymorphisms (SNPs) cut-off, except for rhinovirus (10 SNPs). Patient health records were reviewed for genetically related clusters to identify epidemiological connections.
We collected 436 viral specimens from 359 patients: rhinovirus (n=291), influenza (n=50), HMPV (n=47), and RSV (n=48). Of these, 55% (197/359 patients) were from pediatric hospital and 45% from adult hospitals. Patients ranged in age from 14 days to 93 years, 61% were male, and 74% were white. WGS was performed on 61.2% (178/291) rhinovirus, 78% (39/50) influenza, 92% (44/48) RSV, and all HMPV specimens. Among high-quality genomes, we identified 14 genetically related clusters involving 36 patients, ranging in size from 2-5 patients. We identified common epidemiological links for 53% (19/36) of clustered patients; 63% (12/19) patients had same-unit stay, 26% (5/19) had overlapping hospital stays, and 11% (2/19) shared common provider. On average, genetically related clusters spanned 16 days (range:0-55 days).
WGS offered insights into respiratory virus transmission dynamics. These advancements could potentially improve infection prevention and control strategies, leading to enhanced patient safety and healthcare outcomes.
医疗机构中呼吸道病毒的传播情况尚未得到充分了解。为了调查常见的医疗相关呼吸道病毒感染的传播动态,我们在宾夕法尼亚州匹兹堡的一家儿科医院和两家成人教学医院进行了回顾性全基因组测序(WGS)监测。
从2018年1月2日至2020年1月4日,对住院≥3天的患者的鼻拭子标本进行测序,这些标本对鼻病毒、流感病毒、人偏肺病毒(HMPV)或呼吸道合胞病毒(RSV)呈阳性,采用Illumina平台进行测序。除鼻病毒(10个单核苷酸多态性)外,使用≤3个单核苷酸多态性(SNP)的截断值评估高质量基因组的遗传相关性。查阅患者健康记录以寻找遗传相关的集群,以确定流行病学联系。
我们从359名患者中收集了436份病毒标本:鼻病毒(n = 291)、流感病毒(n = 50)、HMPV(n = 47)和RSV(n = 48)。其中,55%(197/359名患者)来自儿科医院,45%来自成人医院。患者年龄从14天至93岁不等,61%为男性,74%为白人。对61.2%(178/291)的鼻病毒、78%(39/50)的流感病毒、92%(44/48)的RSV以及所有HMPV标本进行了WGS。在高质量基因组中,我们确定了14个遗传相关的集群,涉及36名患者,规模从2至5名患者不等。我们为53%(19/36)的集群患者确定了共同的流行病学联系;63%(12/19)的患者在同一病房住院,26%(5/19)的患者住院时间有重叠,11%(2/19)的患者有共同的医护人员。平均而言,遗传相关的集群跨度为16天(范围:0至55天)。
WGS为呼吸道病毒传播动态提供了见解。这些进展可能会改善感染预防和控制策略,从而提高患者安全性和医疗结果。