Swanson Molly E V, Tan Adelie Y S, Tippett Lynette J, Turner Clinton P, Curtis Maurice A, Scotter Emma L, Lashuel Hilal A, Dragunow Mike, Faull Richard L M, Murray Helen C, Singh-Bains Malvindar K
Centre for Brain Research, University of Auckland, Auckland, New Zealand.
School of Biological Sciences, University of Auckland, Auckland, New Zealand.
Sci Rep. 2025 May 3;15(1):15546. doi: 10.1038/s41598-025-00465-w.
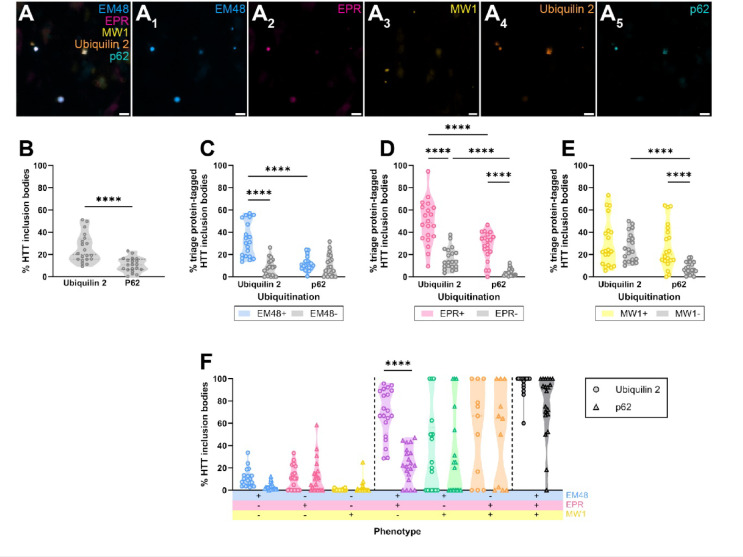
Huntington's disease (HD) is a hereditary neurodegenerative condition caused by a CAG repeat expansion mutation in the gene encoding the huntingtin (HTT) protein. The accumulation of HTT inclusion bodies is a pathological hallmark of HD and a common target for therapeutic strategies. However, the limited efficacy of treatments targeting the HTT protein highlights the need for a better understanding of the role of HTT inclusion bodies in HD pathogenesis. This study examined the heterogeneity of HTT inclusion body composition by co-labelling with three HTT epitope-specific antibodies to characterize HTT inclusion body 'immunophenotype'. We then characterized the size and sub-cellular location of HTT inclusions with distinct immunophenotypes. Using multiplex immunohistochemistry, we also examined the ubiquitination profile of each immunophenotype. Our findings demonstrate that HTT inclusions have a range of immunophenotypes, with some labelled by only one of the three antibodies and others exhibiting co-labelling by several antibodies, thus demonstrating the heterogeneity in inclusion composition and structure. We outline evidence that inclusion bodies exclusively labelled with the EM48 antibody are small, non-nuclear, and more abundant in HD cases with increased CAG repeat length, higher Vonsattel grade, and earlier age of onset. We also find that HTT inclusion bodies labelled by multiple antibodies are more likely to be ubiquitinated, predominantly by K63- rather than K48-linked ubiquitin, suggesting preferential degradation by autophagy. Lastly, we show that ubiquitinated HTT inclusion bodies are more highly immunoreactive for ubiquilin 2 than p62. Our findings highlight the need for multiple antibodies to capture the full spectrum of HTT pathology in HD and imply that future studies should consider the diversity of inclusion body composition and structure when correlating pathology formation to neurodegeneration, clinical symptoms, or disease severity.
亨廷顿病(HD)是一种遗传性神经退行性疾病,由编码亨廷顿蛋白(HTT)的基因中的CAG重复扩增突变引起。HTT包涵体的积累是HD的病理标志,也是治疗策略的常见靶点。然而,针对HTT蛋白的治疗效果有限,这凸显了更好地理解HTT包涵体在HD发病机制中的作用的必要性。本研究通过与三种HTT表位特异性抗体共同标记来检查HTT包涵体组成的异质性,以表征HTT包涵体的“免疫表型”。然后,我们表征了具有不同免疫表型的HTT包涵体的大小和亚细胞定位。使用多重免疫组织化学,我们还检查了每种免疫表型的泛素化谱。我们的研究结果表明,HTT包涵体具有一系列免疫表型,有些仅被三种抗体之一标记,而其他则表现出几种抗体的共同标记,从而证明了包涵体组成和结构的异质性。我们概述了证据,即仅用EM48抗体标记的包涵体较小,非核性,并且在CAG重复长度增加、Vonsattel分级较高和发病年龄较早的HD病例中更为丰富。我们还发现,被多种抗体标记的HTT包涵体更有可能被泛素化,主要是通过K63连接而不是K48连接的泛素,这表明自噬优先降解。最后,我们表明,泛素化的HTT包涵体对泛素连接蛋白2的免疫反应性比对p62更高。我们的研究结果强调了需要多种抗体来捕捉HD中HTT病理学的全貌,并暗示未来的研究在将病理形成与神经退行性变、临床症状或疾病严重程度相关联时应考虑包涵体组成和结构的多样性。