Fischer Tobias, Gredy Sina, Scheel Nadine, Benz Peter M, Fissler Benjamin, Ullrich Melanie, Abeßer Marco, Rokita Adam G, Reichle Jochen, Maier Lars S, Ritter Oliver, Baba Hideo A, Schuh Kai
Institute of Physiology I, University of Wuerzburg, 97070 Wuerzburg, Germany.
Department of Cardiology and Pneumology, Georg-August-University, 37075 Goettingen, Germany.
Cells. 2025 Apr 28;14(9):645. doi: 10.3390/cells14090645.
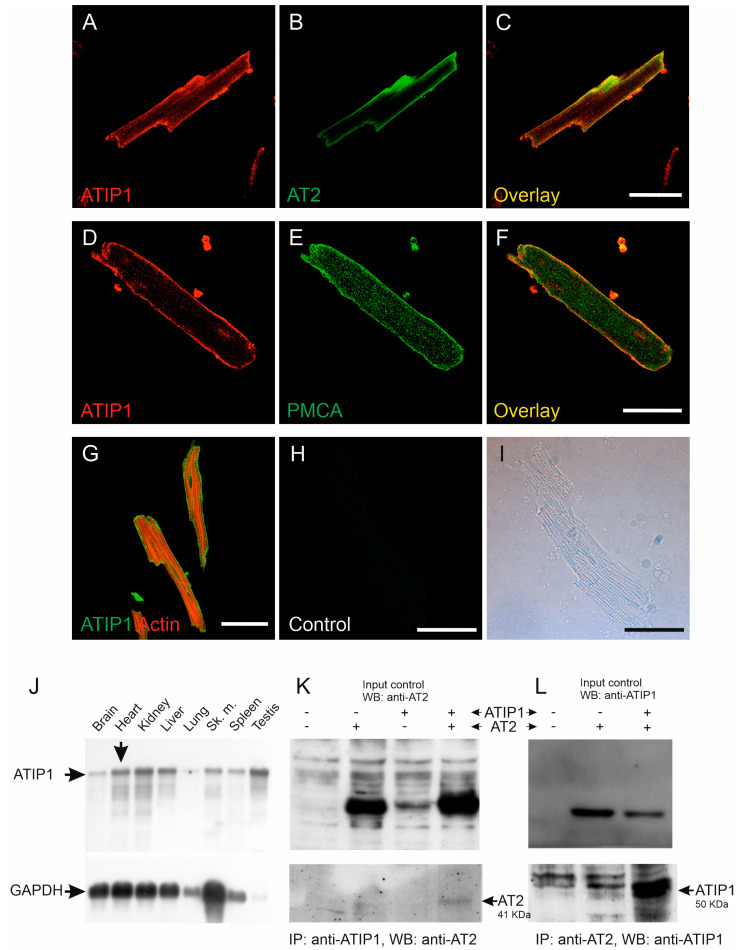
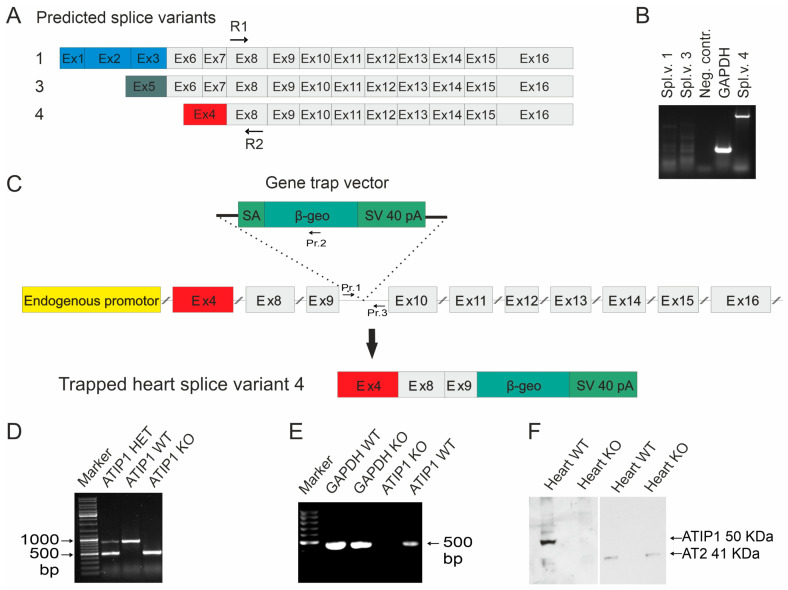
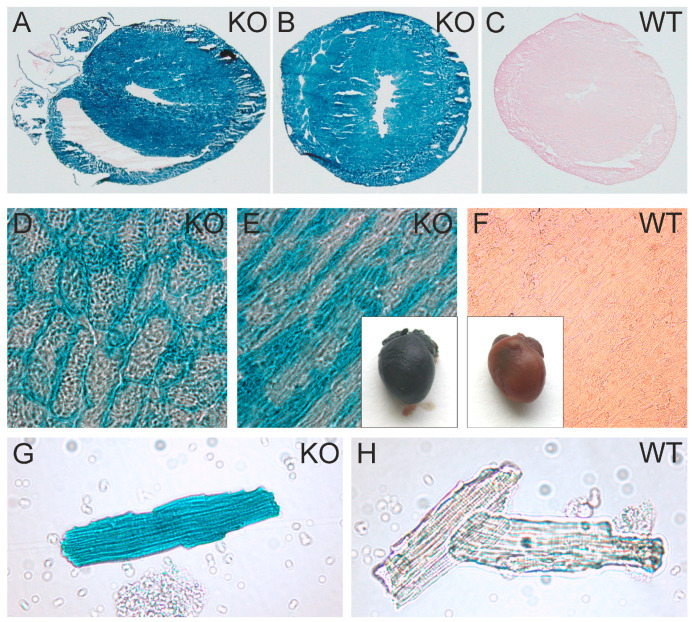
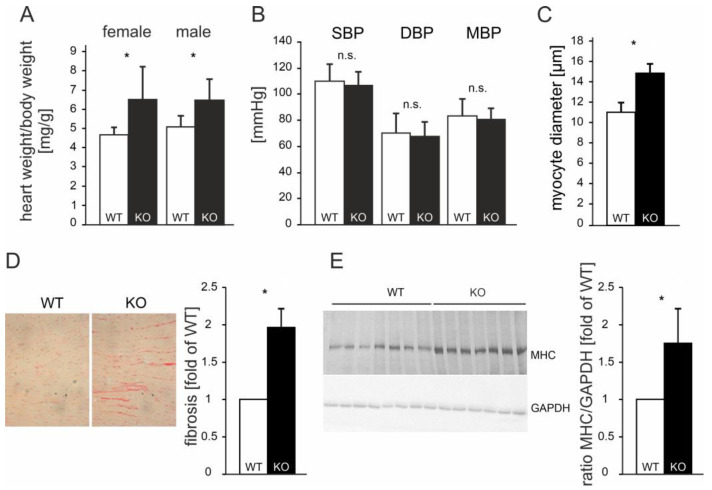
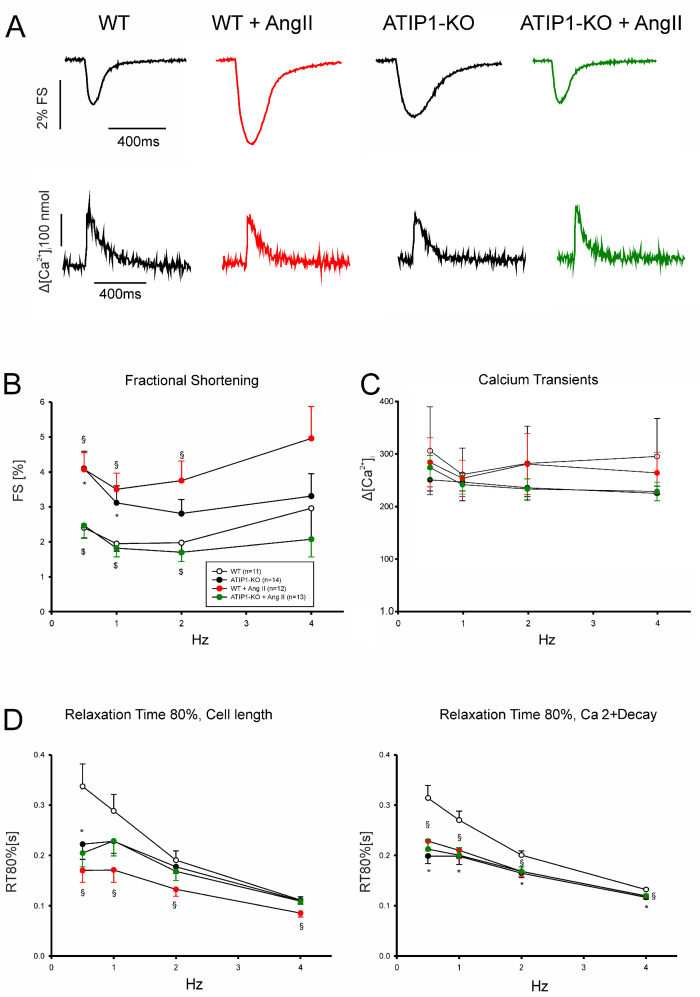
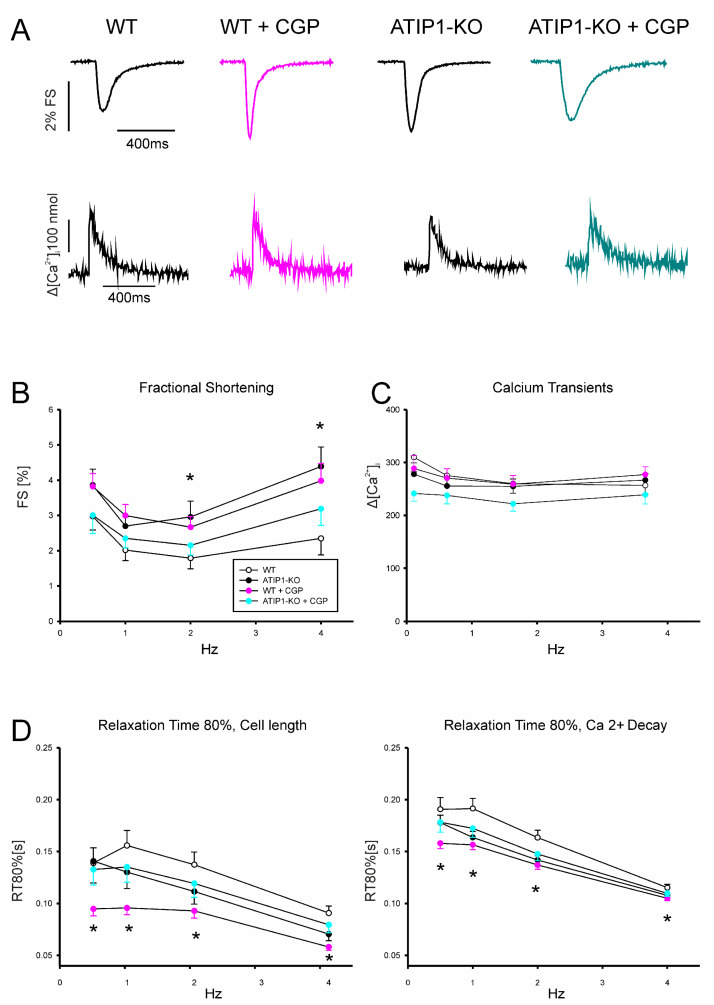
So far, the molecular functions of the angiotensin-type-2 receptor (AT2) interacting protein (ATIP1) have remained unclear, although expression studies have revealed high levels of ATIP1 in the heart. To unravel its physiological function, we investigated ATIP1-KO mice. They develop a spontaneous cardiac hypertrophy with a significantly increased heart/bodyweight ratio, enlarged cardiomyocyte diameters, and augmented myocardial fibrosis. Hemodynamic measurements revealed an increased ejection fraction (EF) in untreated ATIP1-KO mice, and reduced end-systolic and end-diastolic volumes (ESV and EDV), which, in sum, reflect a compensated concentric cardiac hypertrophy. Importantly, no significant differences in blood pressure (BP) were observed. Chronic angiotensin II (AngII) infusion resulted in increases in BP and EF in ATIP1-KO and WT mice. Reductions in ESV and EDV occurred in both ATIP1-KO and WT but to a lesser extent in ATIP1-KOs. Isolated cardiomyocytes exhibited a significantly increased contractility in ATIP1-KO and accelerated Ca decay. AngII treatment resulted in increased fractional shortening in WT but decreased shortening in ATIP1-KO, accompanied by accelerated cell relaxation in WT but absent effects on relaxation in ATIP1-KO cells. The AT2 agonist CGP42112A increased shortening in WT cardiomyocytes but, again, did not affect shortening in ATIP1-KO cells. Relaxation was accelerated by CGP42112A in WT but was unaffected in ATIP1-KO cells. We show that ATIP1 deficiency results in spontaneous cardiac hypertrophy in vivo and that ATIP1 is a downstream signal in the AT2 pathway regulating cell contractility. We hypothesize that the latter effect is because of a disinhibition of the AT1 pathway by impaired AT2 signaling.
到目前为止,血管紧张素2型受体(AT2)相互作用蛋白(ATIP1)的分子功能仍不清楚,尽管表达研究显示心脏中ATIP1水平很高。为了阐明其生理功能,我们对ATIP1基因敲除小鼠进行了研究。它们会自发出现心脏肥大,心脏/体重比显著增加,心肌细胞直径增大,心肌纤维化加剧。血流动力学测量显示,未经治疗的ATIP1基因敲除小鼠射血分数(EF)增加,收缩末期和舒张末期容积(ESV和EDV)减小,总体反映为代偿性向心性心脏肥大。重要的是,未观察到血压(BP)有显著差异。慢性输注血管紧张素II(AngII)导致ATIP1基因敲除小鼠和野生型小鼠的血压和EF升高。ATIP1基因敲除小鼠和野生型小鼠的ESV和EDV均降低,但ATIP1基因敲除小鼠降低程度较小。分离的心肌细胞在ATIP1基因敲除小鼠中表现出收缩力显著增加和钙衰减加速。AngII处理导致野生型小鼠的缩短分数增加,但ATIP1基因敲除小鼠的缩短分数降低,同时野生型小鼠细胞松弛加速,而对ATIP1基因敲除细胞的松弛无影响。AT2激动剂CGP42112A增加了野生型心肌细胞的缩短,但同样对ATIP1基因敲除细胞的缩短无影响。CGP42112A使野生型小鼠细胞松弛加速,但对ATIP1基因敲除细胞无影响。我们发现,ATIP1缺乏会导致体内自发心脏肥大,且ATIP1是AT2途径中调节细胞收缩力的下游信号。我们推测,后一种效应是由于AT2信号受损导致AT1途径去抑制所致。