Uwibambe Esther, Yalcouyé Abdoulaye, Aboagye Elvis Twumasi, Xhakaza Lettilia, Popel Kalinka, Dukuze Norbert, Bharadwaj Thashi, de Kock Carmen, Schrauwen Isabelle, Leal Suzanne M, Mutesa Leon, Wonkam Ambroise
Center for Human Genetics, College of Medicine and Health Sciences, University of Rwanda, Kigali, Rwanda.
Division of Human Genetics, Department of Medicine, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa.
BMC Med Genomics. 2025 May 13;18(1):85. doi: 10.1186/s12920-025-02153-0.
In 30% of patients who exhibit the clinical profile of Cornelia de Lange Syndrome (CdLS), the genetic cause remains undetermined. This proportion tends to be higher in low-resource settings including Africa. We performed a molecular characterization of CdLS in a multiplex Rwandan family.
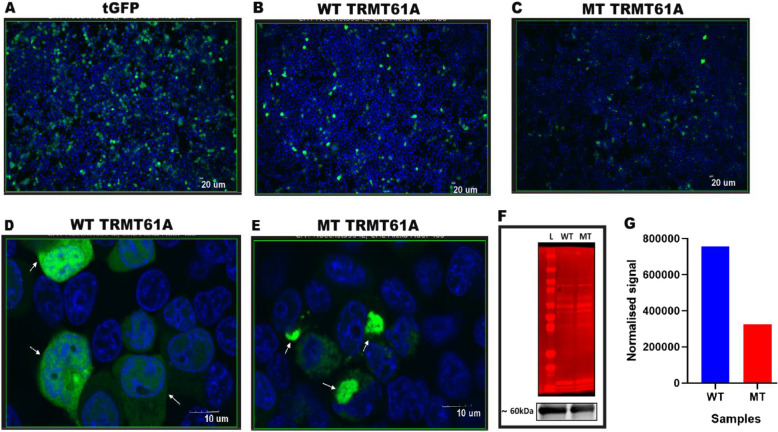
After a clinical evaluation of two affected siblings, DNA isolated from peripheral whole blood of the affected patients and their parents underwent Exome Sequencing (ES). Sanger sequencing validated the variant segregating with CdLS. In silico predictive tools, protein modelling, and cell-based experiments using HEK293T cells were used to investigate the pathogenicity of the variant found.
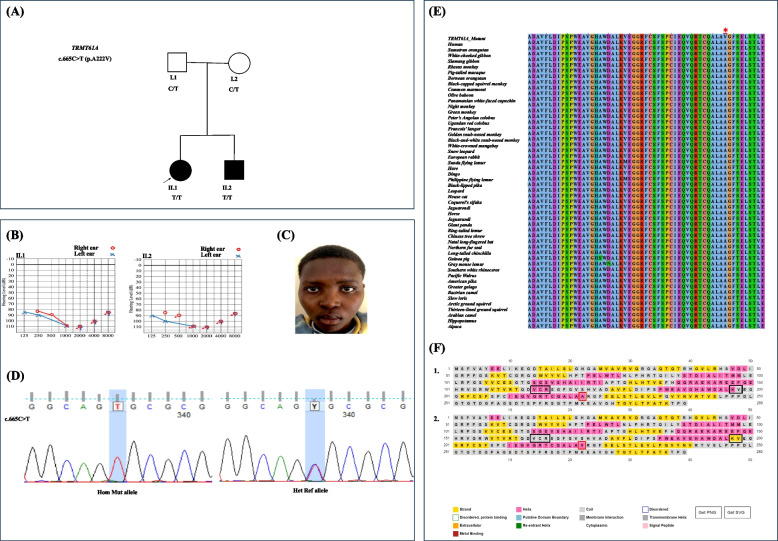
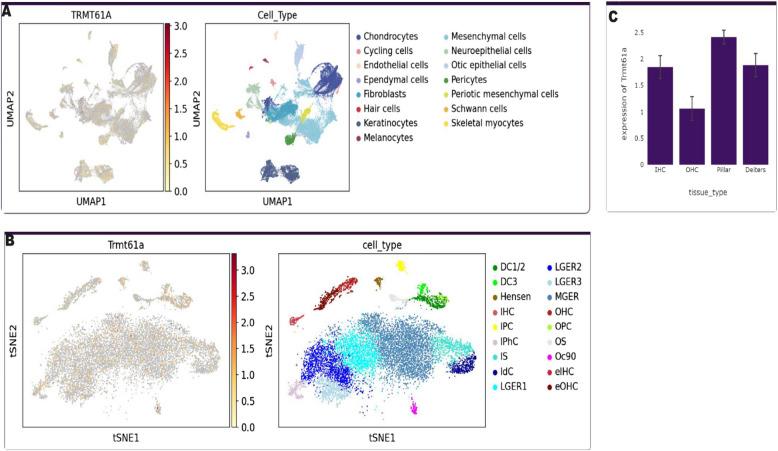
We identified a family with two parents and their two offspring (male and female), who were referred for hearing impairment. The 17-year-old female presented bilateral profound hearing impairment with moderate hypertelorism, progressive visual impairment, and secondary amenorrhea. The 14-year-old male displayed intellectual disability and a bilateral profound hearing impairment with no noticeable facial dysmorphism. Following exome sequencing (ES) of DNA samples obtained from the four family members, we found that the siblings harbored a novel likely pathogenic homozygous missense variant in the TRMT61 A gene [NM_152307.3:c.665C > T p.(Ala222Val)] inherited from both heterozygous parents. In silico analysis suggested that the variant substitutes a highly conserved amino acid, and 2-D structure modelling revealed a significant decrease in the stability of the protein. Cell-based experiment in HEK293T showed that the variant significantly affected the TRMT61 A protein localization which is thought to impact the mitochondrial and cytosolic functions.
We reported a novel biallelic variant in TRMT61 A, [NM_152307.3:c.665C > T p.(Ala222Val)], which is associated with autosomal recessive atypical CdLS in a multiplex Rwandan family, the first report from Africa, and the second globally. The study emphasizes the need to expand the availability of ES for molecular characterization of rare diseases for the understudied genetically diverse population of Africa.
在表现出科妮莉亚·德朗热综合征(CdLS)临床特征的患者中,30%的患者遗传病因仍未明确。在包括非洲在内的资源匮乏地区,这一比例往往更高。我们对一个卢旺达大家庭中的CdLS进行了分子特征分析。
在对两名患病兄弟姐妹进行临床评估后,从患病患者及其父母的外周全血中分离出的DNA进行了外显子组测序(ES)。桑格测序验证了与CdLS共分离的变异。使用计算机预测工具、蛋白质建模以及利用HEK293T细胞进行的基于细胞的实验,来研究发现的变异的致病性。
我们鉴定出一个有两名父母及其两名后代(一男一女)的家庭,他们因听力障碍前来就诊。17岁的女性表现为双侧重度听力障碍,伴有中度眼距增宽、进行性视力损害和继发性闭经。14岁的男性表现为智力残疾和双侧重度听力障碍,无明显面部畸形。对从这四名家庭成员获取的DNA样本进行外显子组测序(ES)后,我们发现这两名兄弟姐妹在TRMT61A基因中携带一种新的可能致病的纯合错义变异[NM_152307.3:c.665C>T p.(Ala222Val)],该变异遗传自双亲杂合子。计算机分析表明,该变异替代了一个高度保守的氨基酸,二维结构建模显示蛋白质稳定性显著降低。在HEK293T细胞中进行的基于细胞的实验表明,该变异显著影响TRMT61A蛋白定位,而这被认为会影响线粒体和胞质功能。
我们报告了TRMT61A基因中的一种新的双等位基因变异[NM_152307.3:c.665C>T p.(Ala222Val)],它与一个卢旺达大家庭中的常染色体隐性非典型CdLS相关,这是来自非洲的首例报告,也是全球第二例。该研究强调了有必要扩大外显子组测序的可及性,以便对非洲研究不足的遗传多样性人群中的罕见疾病进行分子特征分析。