Guo Yang, Pernal Katarzyna
School of Chemistry and Chemical Engineering, Shandong University, Qingdao 266237, Shandong, China.
Institute of Physics, Lodz University of Technology, ul. Wolczanska 217/221, Lodz 93-005, Poland.
J Chem Theory Comput. 2025 Jun 10;21(11):5545-5558. doi: 10.1021/acs.jctc.5c00582. Epub 2025 May 28.
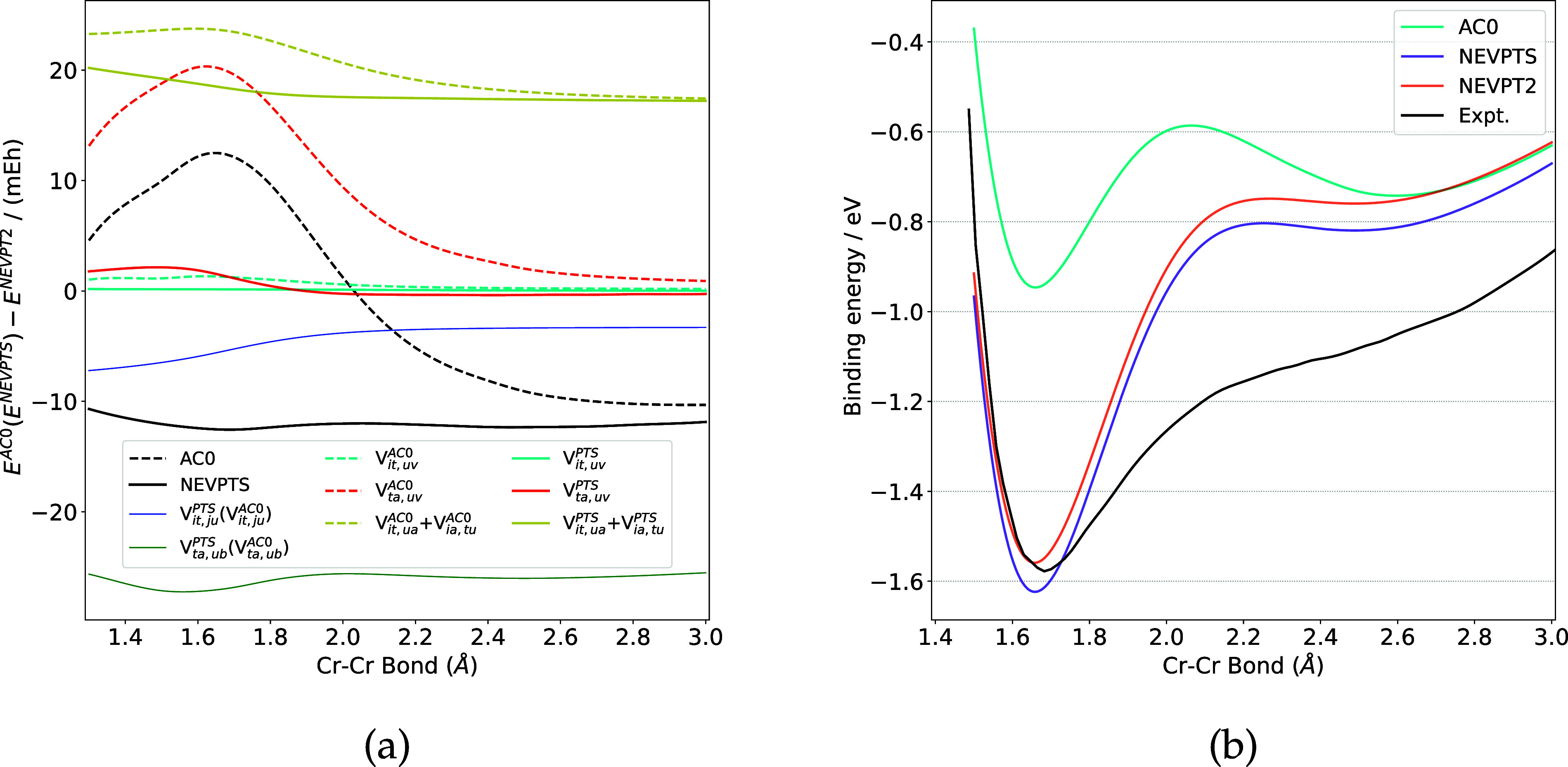
Inspired by the linearized adiabatic connection (AC0) theory, an approximation to second-order N-electron valence state perturbation theory (NEVPT2) has been developed, termed NEVPT within singles (NEVPTS). This approach utilizes amplitudes derived from approximate single-excitation wave functions, requiring only 3rd-order reduced density matrices (RDMs). Consequently, it avoids the computational bottleneck associated with the construction of 4th-order RDMs in NEVPT2. The NEVPTS method demonstrates comparable performance to NEVPT2 in describing potential energy curves for diatomic molecules and singlet-triplet gaps in biradicals, while achieving superior accuracy to AC0 in these applications. For excitation energies of organic molecules, NEVPTS is less accurate than NEVPT2. The overall performance and computational costs of the NEVPTS method lie between those of NEVPT2 and AC0.
受线性化绝热连接(AC0)理论的启发,人们开发了一种二阶N电子价态微扰理论(NEVPT2)的近似方法,称为单重态内的NEVPT(NEVPTS)。该方法利用从近似单激发波函数导出的振幅,仅需要三阶约化密度矩阵(RDM)。因此,它避免了与NEVPT2中构建四阶RDM相关的计算瓶颈。在描述双原子分子的势能曲线和双自由基中的单重态-三重态能隙时,NEVPTS方法表现出与NEVPT2相当的性能,同时在这些应用中比AC0具有更高的精度。对于有机分子的激发能,NEVPTS的精度低于NEVPT2。NEVPTS方法的整体性能和计算成本介于NEVPT2和AC0之间。