González-Casimiro Carlos M, Cámara-Torres Patricia, Merino Beatriz, Astudillo Alma M, de la Fuente Miguel A, Ramírez Cristina M, Alonso Andrés, Cózar-Castellano Irene, Perdomo Germán
Instituto de Biomedicina y Genética Molecular (Consejo Superior de Investigaciones Científicas-Universidad de Valladolid), c/ Sanz y Forés, 3, 47003, Valladolid, Spain.
Departamento de Biología Celular, Genética, Histología y Farmacología, Facultad de Medicina, Universidad de Valladolid, 47002, Valladolid, Spain.
Sci Rep. 2025 May 31;15(1):19168. doi: 10.1038/s41598-025-03790-2.
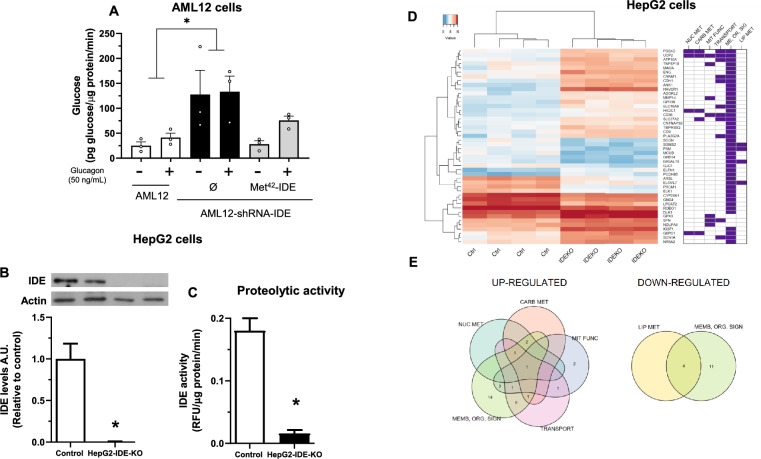
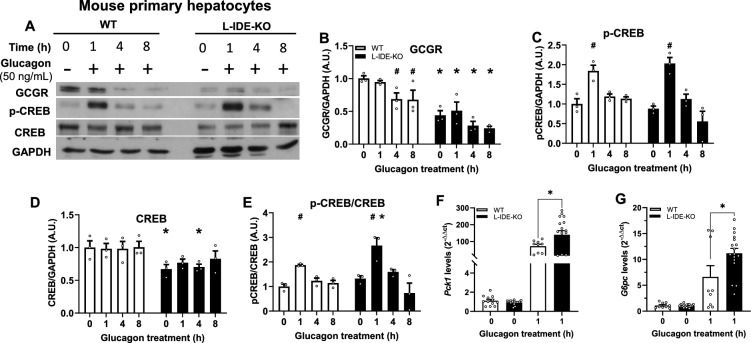
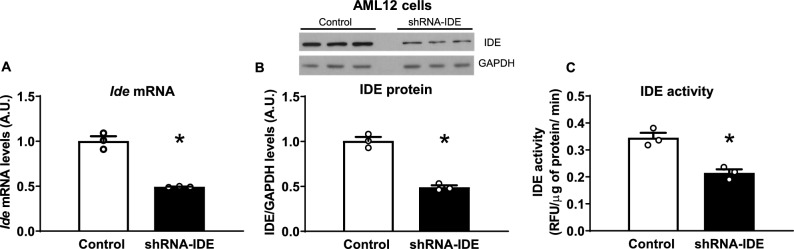
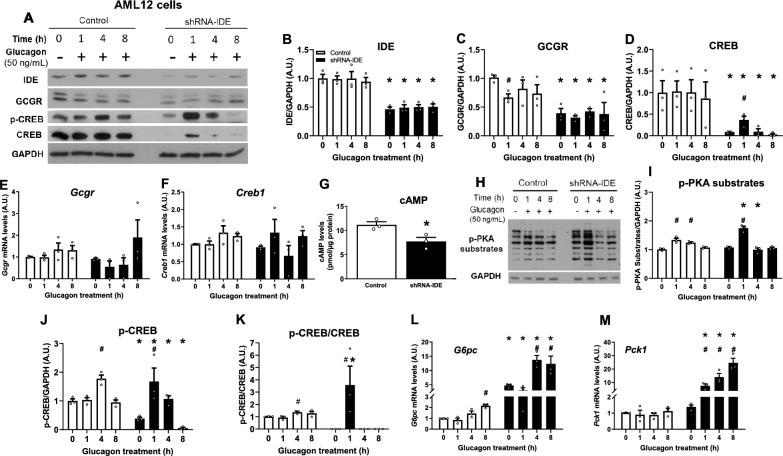
Insulin-degrading enzyme (IDE) is a protein with proteolytic and non-proteolytic functions that regulates glucose homeostasis. In the fasted state, glucagon regulates glycemia through induction of hepatic gluconeogenesis. The rate of hepatic gluconeogenesis is elevated in subjects with type 2 diabetes (T2D) compared with healthy subjects. Interestingly, subjects with T2D show decreased expression of hepatic IDE. However, the role of IDE on the regulation of hepatic gluconeogenesis is completely unknow. We hypothesize that IDE deficiency alters glucagon signaling and thereby gluconeogenesis. To test this hypothesis, we used mouse liver tissues and cultured hepatocytes with total or partial IDE deficiency. The glucagon signaling pathway, expression of gluconeogenic genes, glucose production, and transcriptomic analysis were performed in control and IDE-KO hepatocytes. Total or partial loss of IDE in liver tissues or cultured mouse hepatocytes resulted in lower levels of the glucagon receptor (GCGR) and the cAMP-response element binding protein (CREB). However, glucagon stimulation increased the phosphorylation of CREB, despite lower levels of cAMP in IDE-deficient mouse hepatocytes. The activation of CREB was associated with an upregulation of the gluconeogenic genes Pck1 and G6pc (~ 200% and ~ 70% respectively) and higher glucose production in IDE-deficient mouse hepatocytes. Finally, genetic depletion of IDE in HepG2 hepatocytes led to upregulation of genes involved in cellular functions related to membranes, organelles and signaling receptors. These findings may be of relevance to better understand the regulation of hepatic gluconeogenesis and the use of IDE as a potential therapeutic target for the treatment of T2D.
胰岛素降解酶(IDE)是一种具有蛋白水解和非蛋白水解功能的蛋白质,可调节葡萄糖稳态。在禁食状态下,胰高血糖素通过诱导肝脏糖异生作用来调节血糖水平。与健康受试者相比,2型糖尿病(T2D)患者的肝脏糖异生速率升高。有趣的是,T2D患者肝脏IDE的表达降低。然而,IDE在肝脏糖异生调节中的作用完全未知。我们假设IDE缺乏会改变胰高血糖素信号传导,从而影响糖异生。为了验证这一假设,我们使用了完全或部分缺乏IDE的小鼠肝脏组织和培养的肝细胞。在对照和IDE基因敲除(IDE-KO)的肝细胞中进行了胰高血糖素信号通路、糖异生基因的表达、葡萄糖生成和转录组分析。肝脏组织或培养的小鼠肝细胞中IDE的完全或部分缺失导致胰高血糖素受体(GCGR)和环磷酸腺苷反应元件结合蛋白(CREB)水平降低。然而,尽管IDE缺乏的小鼠肝细胞中cAMP水平较低,但胰高血糖素刺激仍增加了CREB的磷酸化。CREB的激活与IDE缺乏的小鼠肝细胞中糖异生基因Pck1和G6pc的上调(分别约为200%和70%)以及更高的葡萄糖生成有关。最后,HepG2肝细胞中IDE的基因缺失导致与膜、细胞器和信号受体相关的细胞功能基因上调。这些发现可能有助于更好地理解肝脏糖异生的调节以及将IDE作为治疗T2D的潜在治疗靶点的应用。