Clark Jennifer A, Douglas Jack F
Materials Science and Engineering Division, Material Measurement Laboratory, National Institute of Standards and Technology, Gaithersburg, Maryland20899, United States.
J Phys Chem B. 2025 Jul 3;129(26):6548-6560. doi: 10.1021/acs.jpcb.5c01009. Epub 2025 Jun 3.
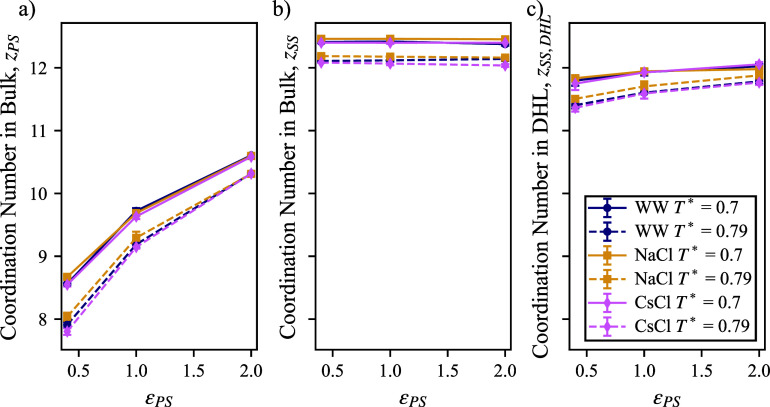
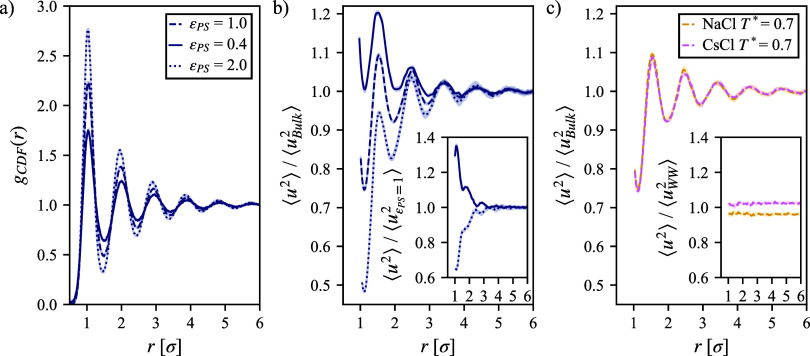
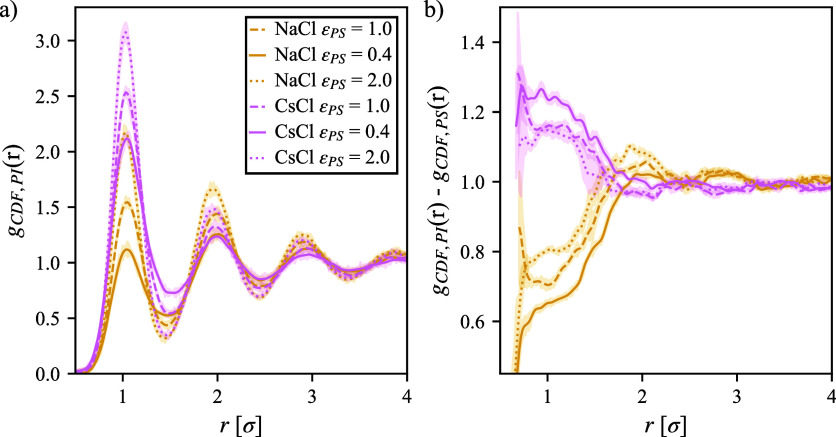
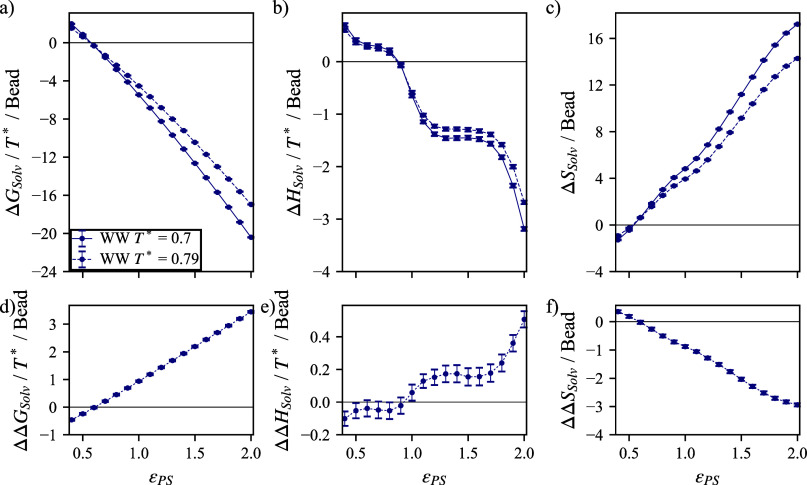
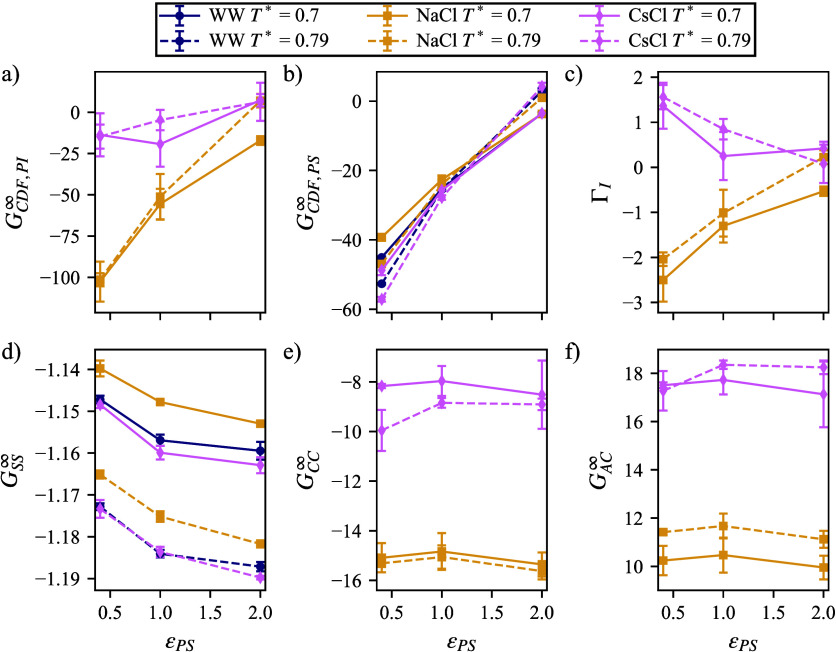
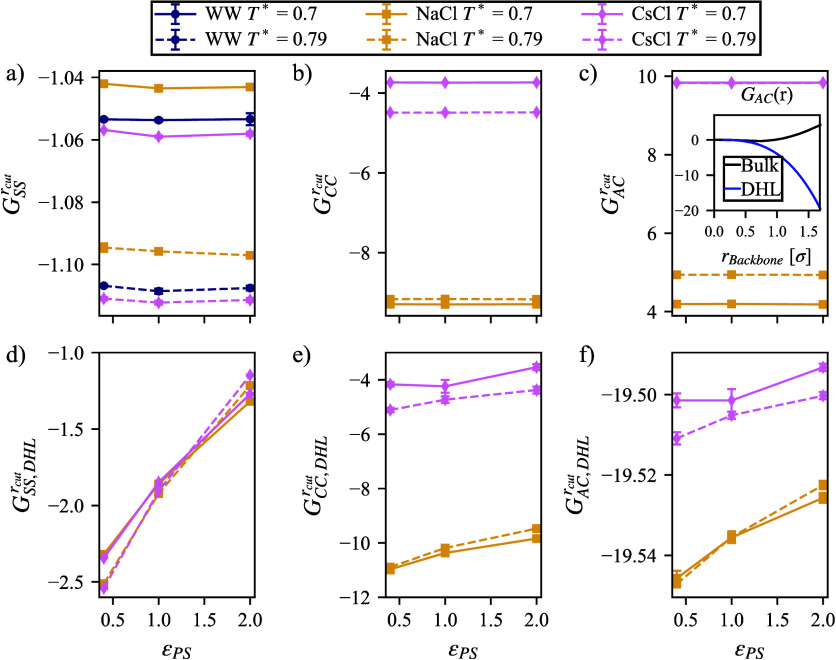
We study the solvation free energy of model water-soluble polymers with an emphasis on better understanding the entropic contribution deriving from the formation of a dynamic hydration layer (DHL). To isolate the solvation free energy due to polymer hydration from contributions that arise from changes in the polymer conformation (and thus solvent-accessible surface area) that ordinarily accompany solvation, we restrict a polymer chain in a rod-like configuration. As in recent works, the nanoscale mobility gradient around the polymer chain, defining the DHL, is quantified through the determination of the Debye-Waller parameter, ⟨⟩, for solvent in the vicinity of the polymer. This gradient enables us to easily visualize the DHL around the polymer. Direct computation of the free energy of solvation indicates a large entropic contribution that correlates with changes in Kirkwood-Buff integrals, which allow us to quantify specific ion effects on polymer solvation. While the water mobility exhibits a significant dependence on the strength of the polymer-solvent interaction in the nanoscale DHL, we unexpectedly found no additional specific ion effect on the mobility within the DHL relative to the bulk solution and, moreover, we find no change in the spatial extent of the DHL to within experimental uncertainty. On the other hand, we find an excess density of CsCl close to the polymer and density depletion of NaCl, consistent with previous suggestions that chaotropic ions partition toward polymer interfaces. Our work indicates that polymer hydration can make a large contribution to polymer solvation free energy, and we expect this phenomenon to be important in relation to understanding the thermodynamics of molecular self-assembly and phase separation processes of water-soluble polymers.
我们研究了模型水溶性聚合物的溶剂化自由能,重点是更好地理解动态水化层(DHL)形成所产生的熵贡献。为了将聚合物水化引起的溶剂化自由能与通常伴随溶剂化的聚合物构象变化(以及因此溶剂可及表面积)所产生的贡献区分开来,我们将聚合物链限制在棒状构型。与最近的工作一样,通过测定聚合物附近溶剂的德拜-瓦勒参数〈〉来量化定义DHL的聚合物链周围的纳米级迁移率梯度。这种梯度使我们能够轻松地可视化聚合物周围的DHL。溶剂化自由能的直接计算表明存在与柯克伍德-布夫积分变化相关的大熵贡献,这使我们能够量化特定离子对聚合物溶剂化的影响。虽然水的迁移率在纳米级DHL中对聚合物-溶剂相互作用的强度有显著依赖性,但我们意外地发现相对于本体溶液,DHL内的迁移率没有额外的特定离子效应,而且,我们发现在实验不确定性范围内DHL的空间范围没有变化。另一方面,我们发现靠近聚合物处CsCl的过量密度和NaCl的密度耗尽,这与之前关于离液序列高的离子向聚合物界面分配的建议一致。我们的工作表明聚合物水化对聚合物溶剂化自由能有很大贡献,并且我们预计这种现象对于理解水溶性聚合物的分子自组装和相分离过程的热力学很重要。