Zimin Aleksey V, Puiu Daniela, Pertea Mihaela, Yorke James A, Salzberg Steven L
Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD.
Center for Computational Biology, Johns Hopkins University, Baltimore, MD.
bioRxiv. 2025 May 12:2025.05.07.652745. doi: 10.1101/2025.05.07.652745.
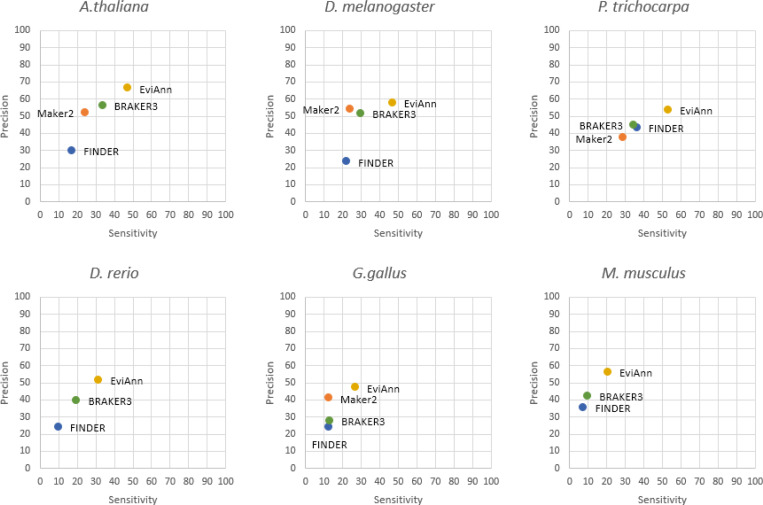
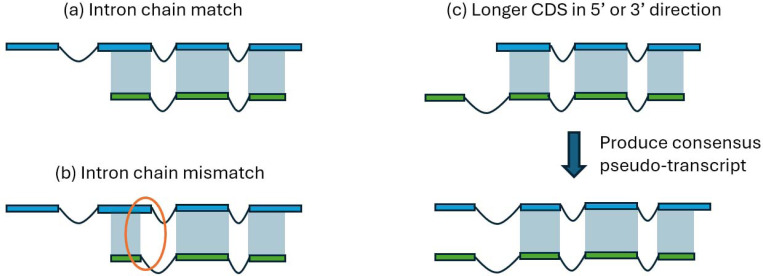
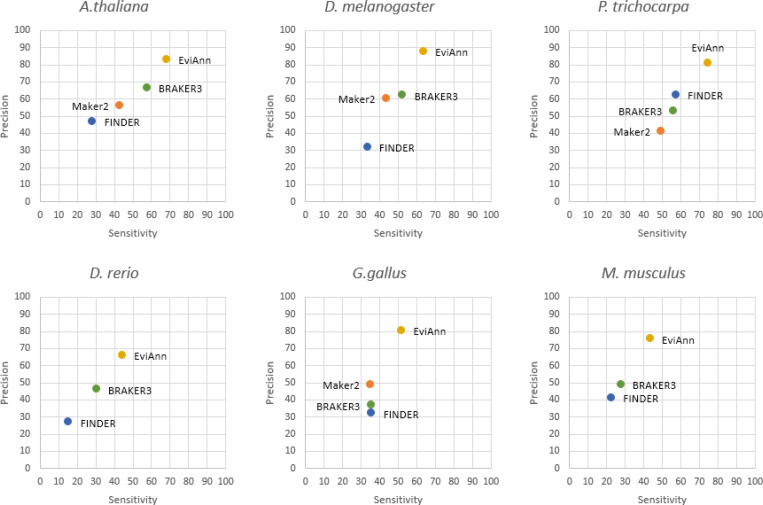
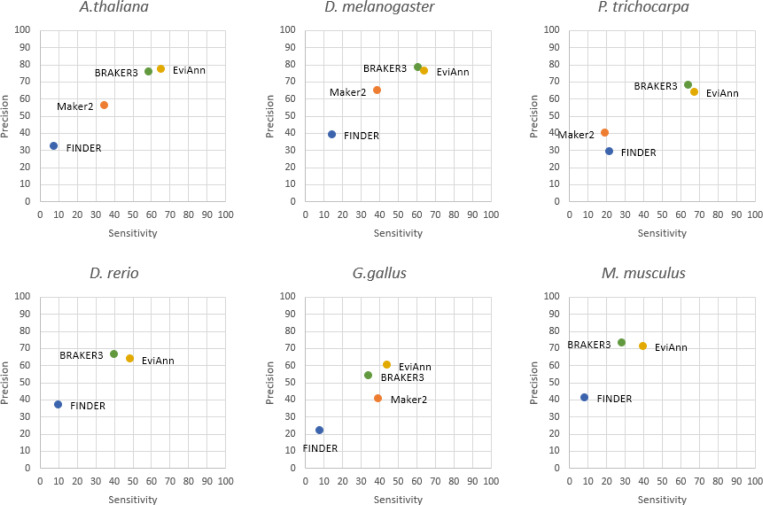
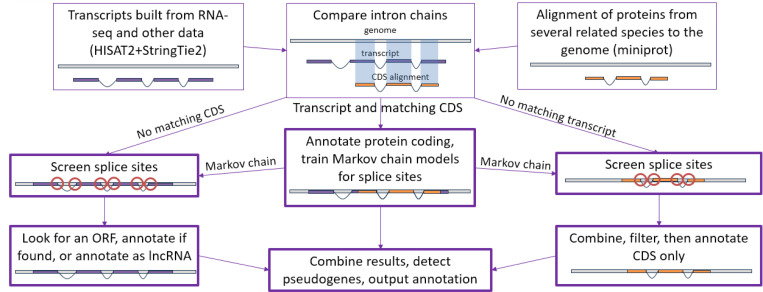
For many years, machine learning-based gene finding approaches have been the central components of eukaryotic genome annotation pipelines, and they remain so today. The reliance on these approaches was originally sustained by the high cost and low availability of gene expression data, a primary source of evidence for gene annotation along with protein homology. However, innovations in modern sequencing technologies have revolutionized the acquisition of abundant gene expression data, allowing us to rely more heavily on this class of evidence. In addition to gene expression data, proteins found in a multitude of well-annotated genomes represent another invaluable resource for gene annotation. Existing annotation packages often underutilize these data sources, which prompted us to develop EviAnn (Evidence-based Annotation), a novel evidence-based eukaryotic gene annotation system. EviAnn takes a strongly data-driven approach, building the exon-intron structure of genes from transcript alignments or protein-sequence homology rather than from purely gene finding techniques. We show that when provided with the same input data, EviAnn consistently outperforms current state-of-the-art packages including BRAKER3, MAKER2, and FINDER, while utilizing considerably less computer time. Annotation of a mammalian genome can be completed in less than an hour on a single multi-core server. EviAnn is freely available under an open-source license from https://github.com/alekseyzimin/EviAnn_release.
多年来,基于机器学习的基因发现方法一直是真核生物基因组注释流程的核心组成部分,如今依然如此。对这些方法的依赖最初是由于基因表达数据的高成本和低可得性,基因表达数据是与蛋白质同源性一起作为基因注释主要证据来源。然而,现代测序技术的创新彻底改变了丰富基因表达数据的获取方式,使我们能够更依赖这类证据。除了基因表达数据,在众多注释良好的基因组中发现的蛋白质是基因注释的另一个宝贵资源。现有的注释软件包常常未充分利用这些数据源,这促使我们开发了EviAnn(基于证据的注释),这是一种新型的基于证据的真核生物基因注释系统。EviAnn采用强烈的数据驱动方法,从转录本比对或蛋白质序列同源性构建基因的外显子 - 内含子结构,而不是纯粹从基因发现技术构建。我们表明,当提供相同的输入数据时,EviAnn始终优于当前最先进的软件包,包括BRAKER3、MAKER2和FINDER,同时使用的计算机时间要少得多。在单个多核服务器上,不到一小时就能完成哺乳动物基因组的注释。EviAnn可根据开源许可从https://github.com/alekseyzimin/EviAnn_release免费获取。