Brůna Tomáš, Lomsadze Alexandre, Borodovsky Mark
School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA.
NAR Genom Bioinform. 2020 Jun;2(2):lqaa026. doi: 10.1093/nargab/lqaa026. Epub 2020 May 13.
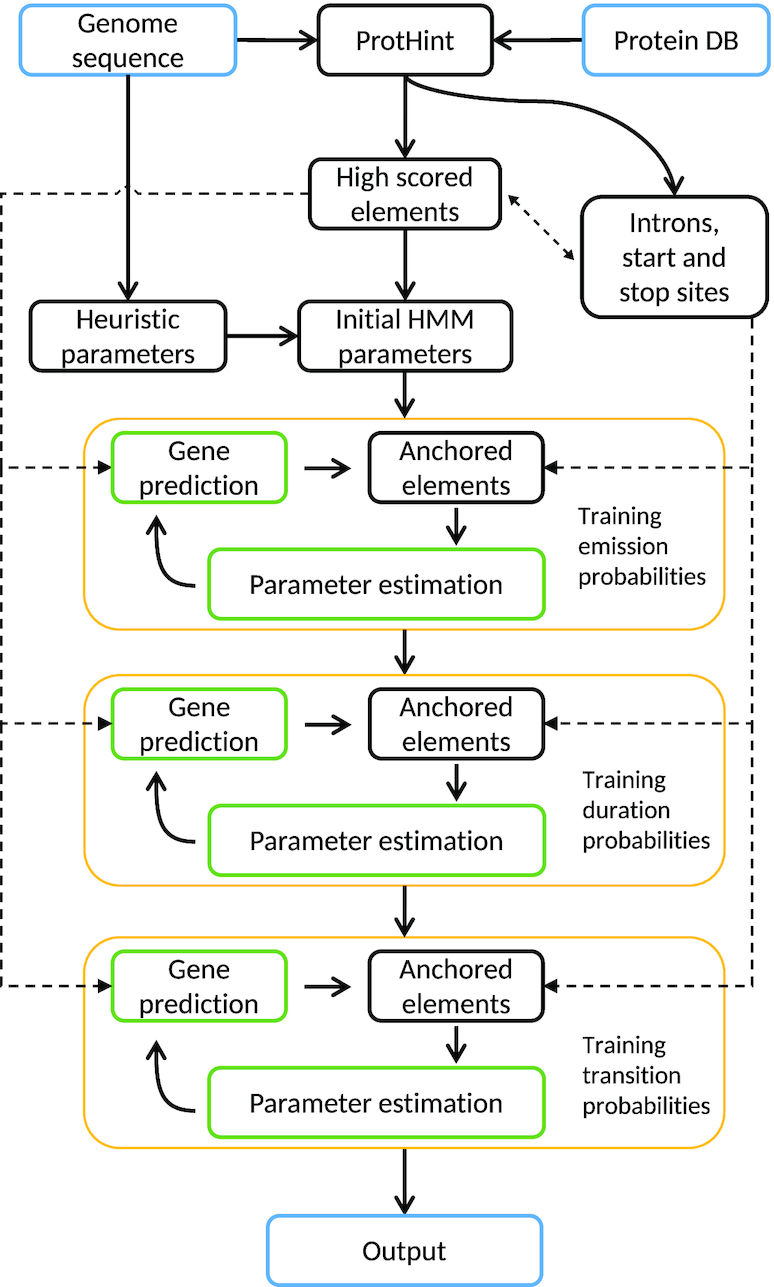
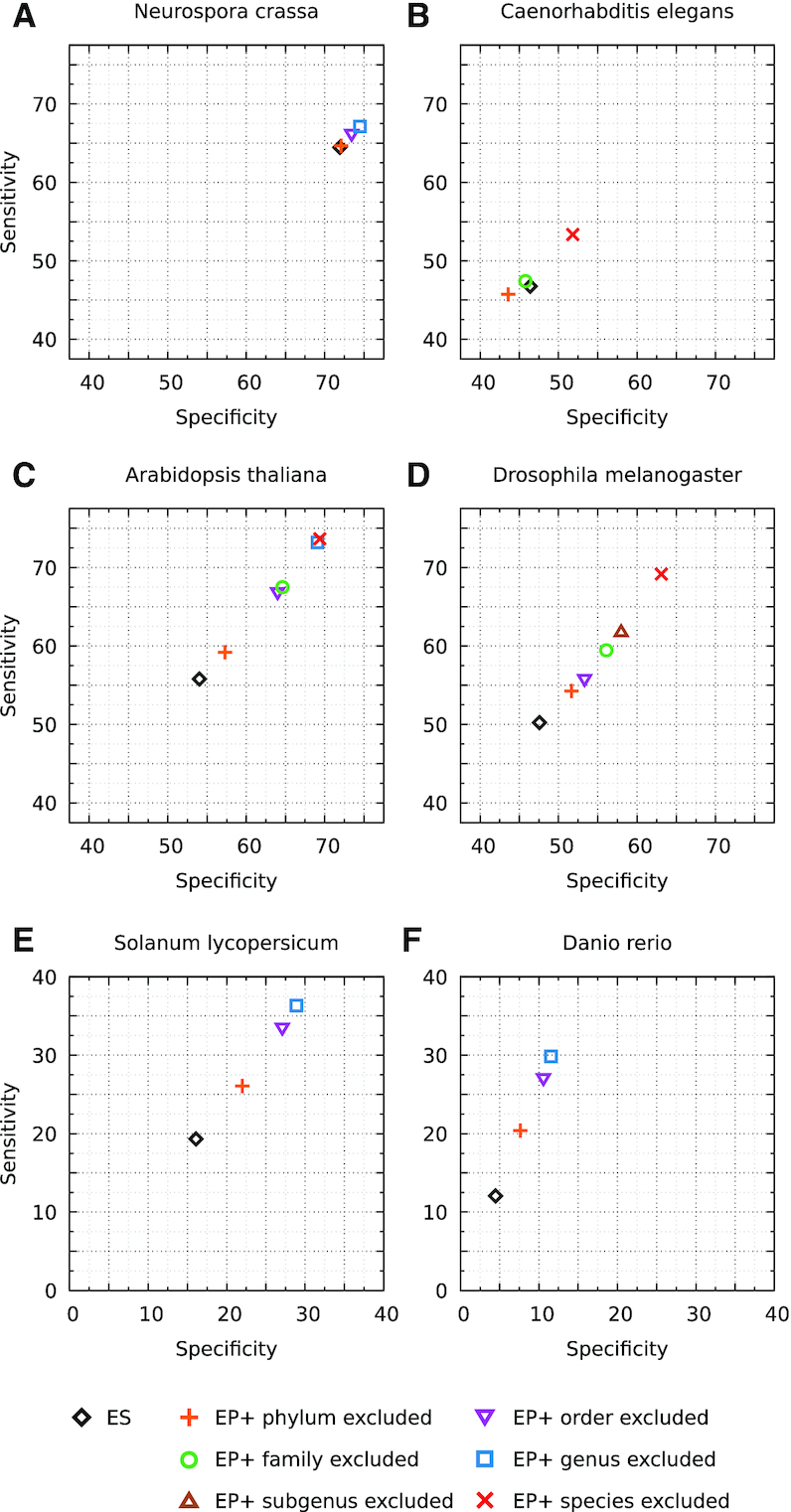
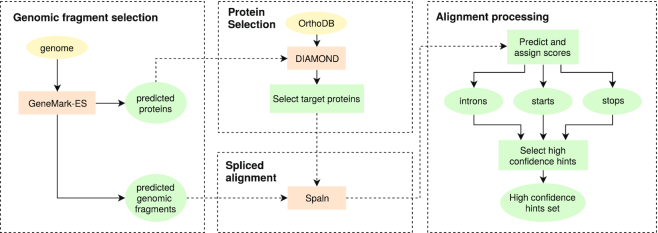
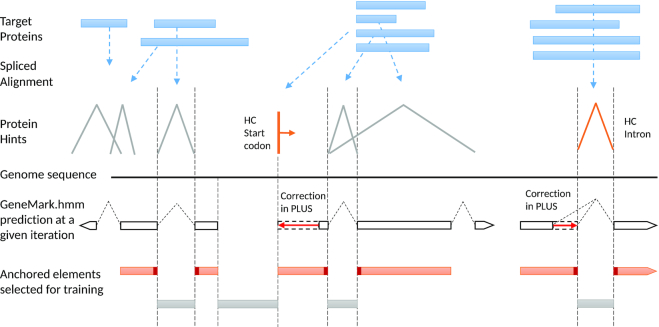
We have made several steps toward creating a fast and accurate algorithm for gene prediction in eukaryotic genomes. First, we introduced an automated method for efficient gene finding, GeneMark-ES, with parameters trained in iterative mode. Next, in GeneMark-ET we proposed a method of integration of unsupervised training with information on intron positions revealed by mapping short RNA reads. Now we describe GeneMark-EP, a tool that utilizes another source of external information, a protein database, readily available prior to the start of a sequencing project. A new specialized pipeline, ProtHint, initiates massive protein mapping to genome and extracts hints to splice sites and translation start and stop sites of potential genes. GeneMark-EP uses the hints to improve estimation of model parameters as well as to adjust coordinates of predicted genes if they disagree with the most reliable hints (the -EP+ mode). Tests of GeneMark-EP and -EP+ demonstrated improvements in gene prediction accuracy in comparison with GeneMark-ES, while the GeneMark-EP+ showed higher accuracy than GeneMark-ET. We have observed that the most pronounced improvements in gene prediction accuracy happened in large eukaryotic genomes.
我们在创建一种用于真核生物基因组基因预测的快速且准确的算法方面已经迈出了几步。首先,我们引入了一种用于高效基因发现的自动化方法——GeneMark-ES,其参数是在迭代模式下训练得到的。接下来,在GeneMark-ET中,我们提出了一种将无监督训练与通过映射短RNA reads揭示的内含子位置信息相结合的方法。现在我们描述GeneMark-EP,这是一种利用另一种外部信息源——蛋白质数据库的工具,该数据库在测序项目开始之前即可获取。一种新的专门流程ProtHint启动对基因组的大规模蛋白质映射,并提取潜在基因的剪接位点以及翻译起始和终止位点的线索。GeneMark-EP利用这些线索来改进模型参数的估计,并且如果预测基因的坐标与最可靠的线索不一致(-EP+模式),还会调整预测基因的坐标。与GeneMark-ES相比,GeneMark-EP和-EP+的测试表明基因预测准确性有所提高,而GeneMark-EP+显示出比GeneMark-ET更高的准确性。我们观察到,基因预测准确性最显著的提高发生在大型真核生物基因组中。