Stephenson Eloise J, Bailey Laura J, Gentleman Stephen, Tuppen Helen, Bodi Istvan, Troakes Claire, Morris Christopher M, Carr Tony M, Guthrie Sarah, Elson Joanna L, Pienaar Ilse S
Department of Neuroscience, School of Life Sciences, University of Sussex, Brighton, UK.
Genome Damage and Stability Centre, School of Life Sciences, University of Sussex, Brighton, UK.
Aging Cell. 2025 Aug;24(8):e70125. doi: 10.1111/acel.70125. Epub 2025 Jun 6.
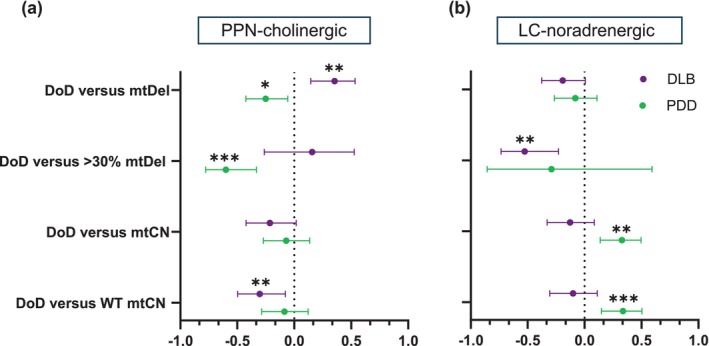
The age-associated neurodegenerative disorder, Lewy body dementia (LBD), encompasses neuropsychiatric symptom-overlapping Dementia with Lewy bodies (DLB) and Parkinson's Disease with Dementia (PDD). We characterised how differential mitochondrial DNA (mtDNA) profiles contribute to neurotype-specific neurodegeneration and thereby clinicopathological heterogeneity, between LBD's syndromes. We further characterised key nuclear-encoding genes' recalibrations in response to such mtDNA changes. In post-mortem 'single-cell' acetylcholine- and noradrenaline-producing neurons, respectively of the pedunculopontine nucleus (PPN) and locus coeruleus (LC) from DLB, PDD and neurological-control brains, we quantified 'major arc'-locating mtDNA deletions (mtDels) and -copy number (mtCN), and measured mRNA levels of nuclear-encoding genes regulating mtDNA maintenance, -biogenesis and mitophagy. DLB cases' OXPHOS defect instigating mtDel burden was higher in both neurotypes than PDD. In DLB, mtCN was reduced for both neurotypes, but PDD cases revealed mtDNA depletion in LC-noradrenergic neurons only. DLB patients' shorter survival correlated with PPN-cholinergic neurons' mtDel levels, inversely with wild-type mtCN, implying that such neurons' inability to maintain sufficient wild-type mtDNA content drive DLBs' rapid psycho-cognitive manifestations. Contrastingly, PDD's longer disease duration allowed compensation against mtDels' clonal expansion in PPN-cholinergic neurons. Moreover, PDD induced mRNA depletion of a mitochondrial genome maintenance gene in PPN-cholinergic neurons, whilst LC-noradrenergic neurons displayed reduced expression of a mitophagy regulating gene. Here we identify mitochondrial genome maintenance and mitophagy pathway enrichment as therapeutic targets to offset defective mtDNA within pontine cholinergic and noradrenergic neurons of PDD patients. The pronounced LBD subtype-related mitochondria-nuclear genetic differences question the consensus that pathology converges at disease end-stage, calling for LBD subtype and neurotype-specific therapeutics.
与年龄相关的神经退行性疾病——路易体痴呆(LBD),包括神经精神症状重叠的路易体痴呆(DLB)和帕金森病痴呆(PDD)。我们研究了不同的线粒体DNA(mtDNA)谱如何导致LBD各综合征之间神经类型特异性神经退行性变,进而导致临床病理异质性。我们进一步研究了关键核编码基因响应此类mtDNA变化的重新校准情况。在死后取自DLB、PDD和神经对照脑的脚桥核(PPN)和蓝斑(LC)的分别产生乙酰胆碱和去甲肾上腺素的“单细胞”神经元中,我们量化了位于“主环”的mtDNA缺失(mtDels)和拷贝数(mtCN),并测量了调节mtDNA维持、生物合成和线粒体自噬的核编码基因的mRNA水平。DLB病例中引发mtDel负荷的氧化磷酸化缺陷在两种神经类型中均高于PDD。在DLB中,两种神经类型的mtCN均降低,但PDD病例仅显示LC去甲肾上腺素能神经元中的mtDNA耗竭。DLB患者较短的生存期与PPN胆碱能神经元的mtDel水平相关,与野生型mtCN呈负相关,这意味着此类神经元无法维持足够的野生型mtDNA含量驱动了DLB快速的心理认知表现。相反,PDD较长的病程允许对PPN胆碱能神经元中mtDels的克隆扩增进行补偿。此外,PDD导致PPN胆碱能神经元中线粒体基因组维持基因的mRNA耗竭,而LC去甲肾上腺素能神经元显示线粒体自噬调节基因的表达降低。在这里,我们确定线粒体基因组维持和线粒体自噬途径富集作为治疗靶点,以抵消PDD患者脑桥胆碱能和去甲肾上腺素能神经元内有缺陷的mtDNA。明显的LBD亚型相关的线粒体-核基因差异对病理学在疾病终末期趋同的共识提出了质疑,这就需要针对LBD亚型和神经类型的特异性治疗方法。