Jaegle Benjamin, Voichek Yoav, Haupt Max, Sotiropoulos Alexandros G, Gauthier Kevin, Heuberger Matthias, Jung Esther, Herren Gerhard, Widrig Victoria, Leber Rebecca, Li Yipu, Schierscher Beate, Serex Sarah, Boczkowska Maja, Jasińska Marta-Puchta, Bolc Paulina, Chalhoub Boulos, Stein Nils, Keller Beat, Sánchez-Martín Javier
Department of Plant and Microbial Biology, University of Zurich, Zollikerstrasse 107, Zurich, 8008, Switzerland.
Gregor Mendel Institute, Austrian Academy of Sciences, Vienna BioCenter, Vienna, 1030, Austria.
Genome Biol. 2025 Jun 18;26(1):172. doi: 10.1186/s13059-025-03645-z.
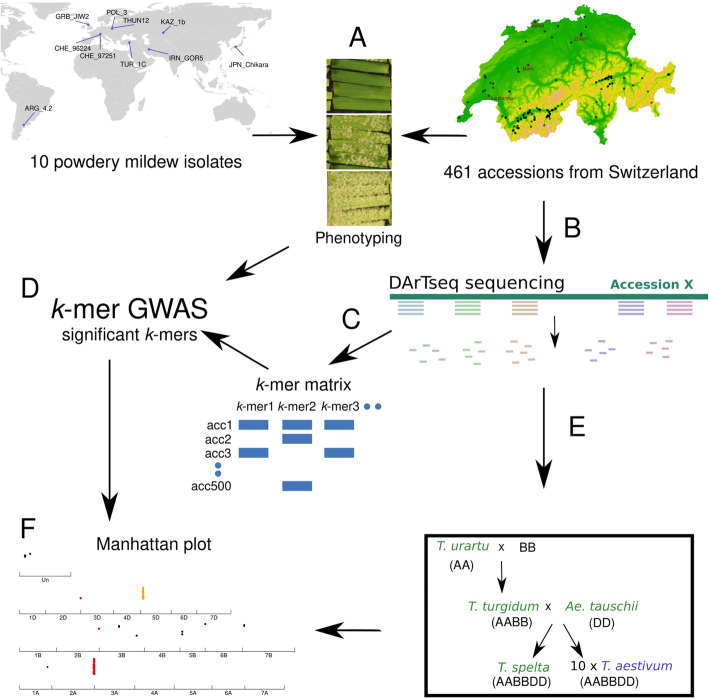
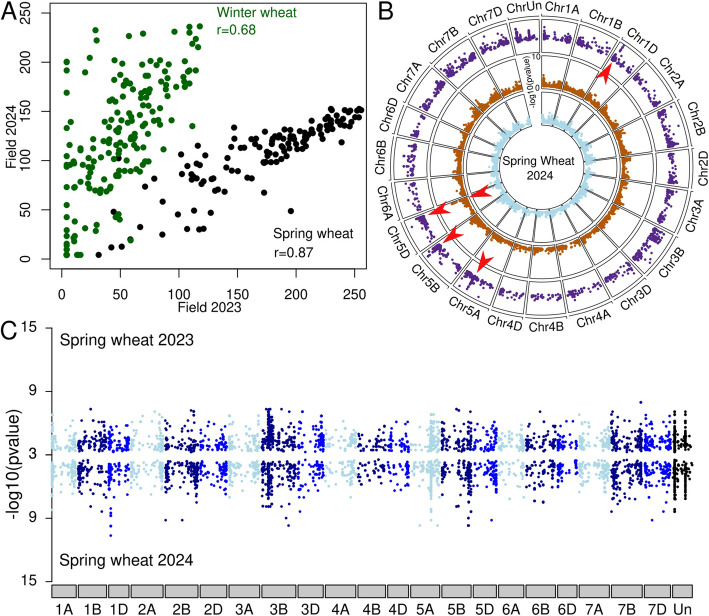
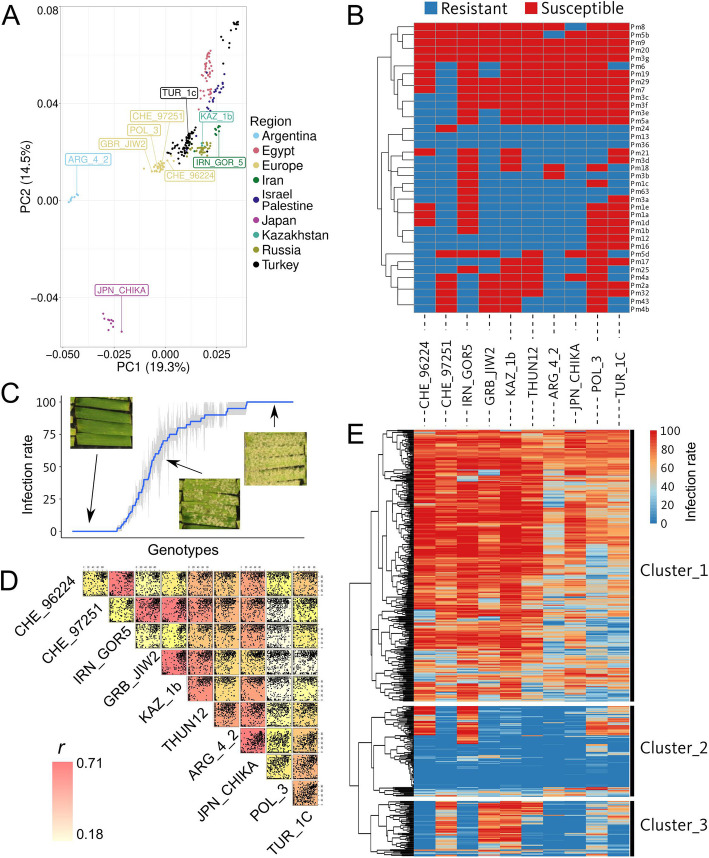
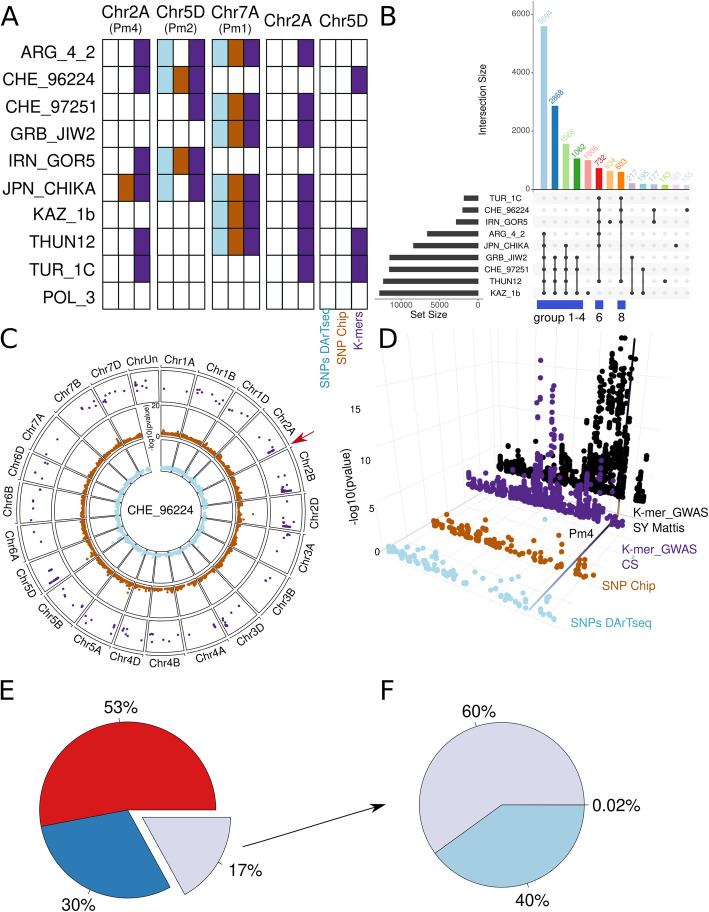
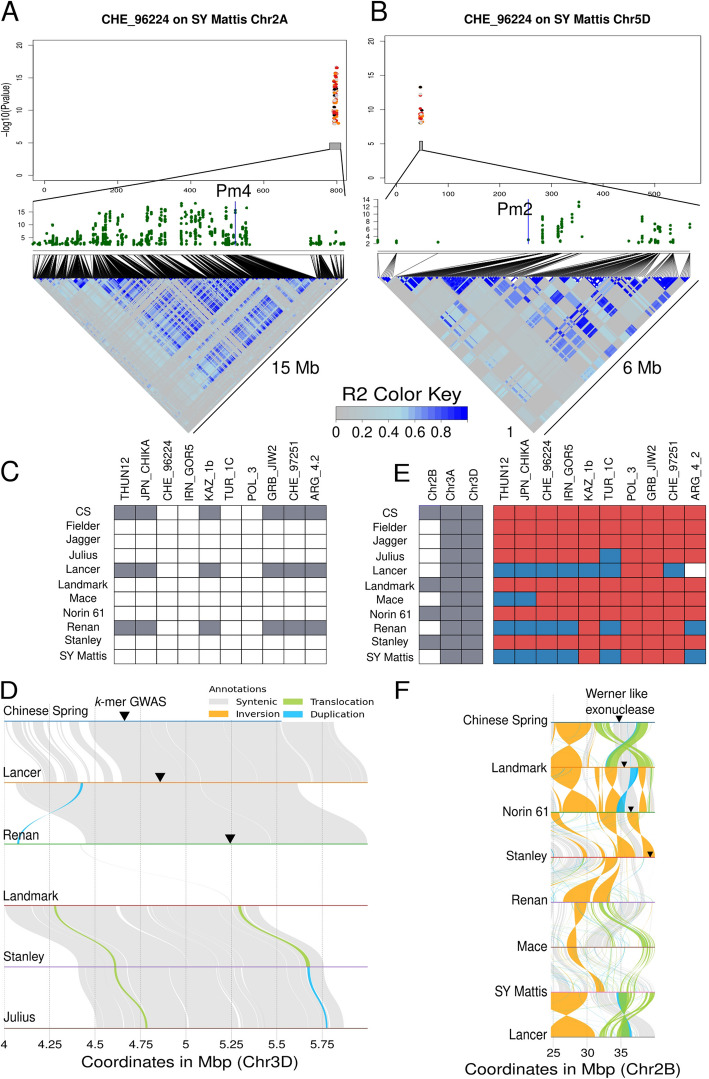
Wheat genetic resources hold the diversity required to mitigate agricultural challenges from climate change and reduced inputs. Using DArTseq, we genotype 461 wheat landraces and cultivars and evaluate them for powdery mildew resistance. By developing a k-mer-based GWAS approach with fully assembled genomes of Triticum aestivum and its progenitors, we uncover 25% more resistance-associated k-mers than single-reference methods, outperforming SNP-based GWAS in both loci detection and mapping precision. In total, we detect 34 powdery mildew resistance loci, including 27 potentially novel regions. Our approach underscores the importance of integrating multiple reference genomes to unlock the potential of wheat germplasm.
小麦遗传资源拥有应对气候变化和减少投入带来的农业挑战所需的多样性。我们利用DArTseq技术对461份小麦地方品种和栽培品种进行基因分型,并评估它们对白粉病的抗性。通过开发一种基于k-mer的全基因组关联研究(GWAS)方法,利用普通小麦及其祖先的全基因组组装序列,我们发现与抗性相关的k-mer比单参考方法多25%,在基因座检测和定位精度方面均优于基于单核苷酸多态性(SNP)的GWAS。我们总共检测到34个白粉病抗性基因座,其中包括27个潜在的新区域。我们的方法强调了整合多个参考基因组以挖掘小麦种质潜力的重要性。