Pannonhalmi Ádám, Sipos Bence, Kurucz Róbert Imre, Katona Gábor, Kemény Lajos, Csóka Ildikó
Department of Dermatology and Allergology, University of Szeged, Korányi Alley 6, H-6720 Szeged, Hungary.
Institute of Pharmaceutical Technology and Regulatory Affairs, University of Szeged, Eötvös Street 6, H-6720 Szeged, Hungary.
Pharmaceuticals (Basel). 2025 Jun 12;18(6):876. doi: 10.3390/ph18060876.
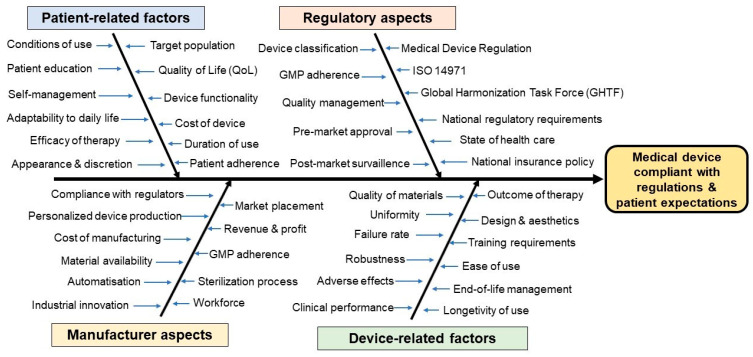
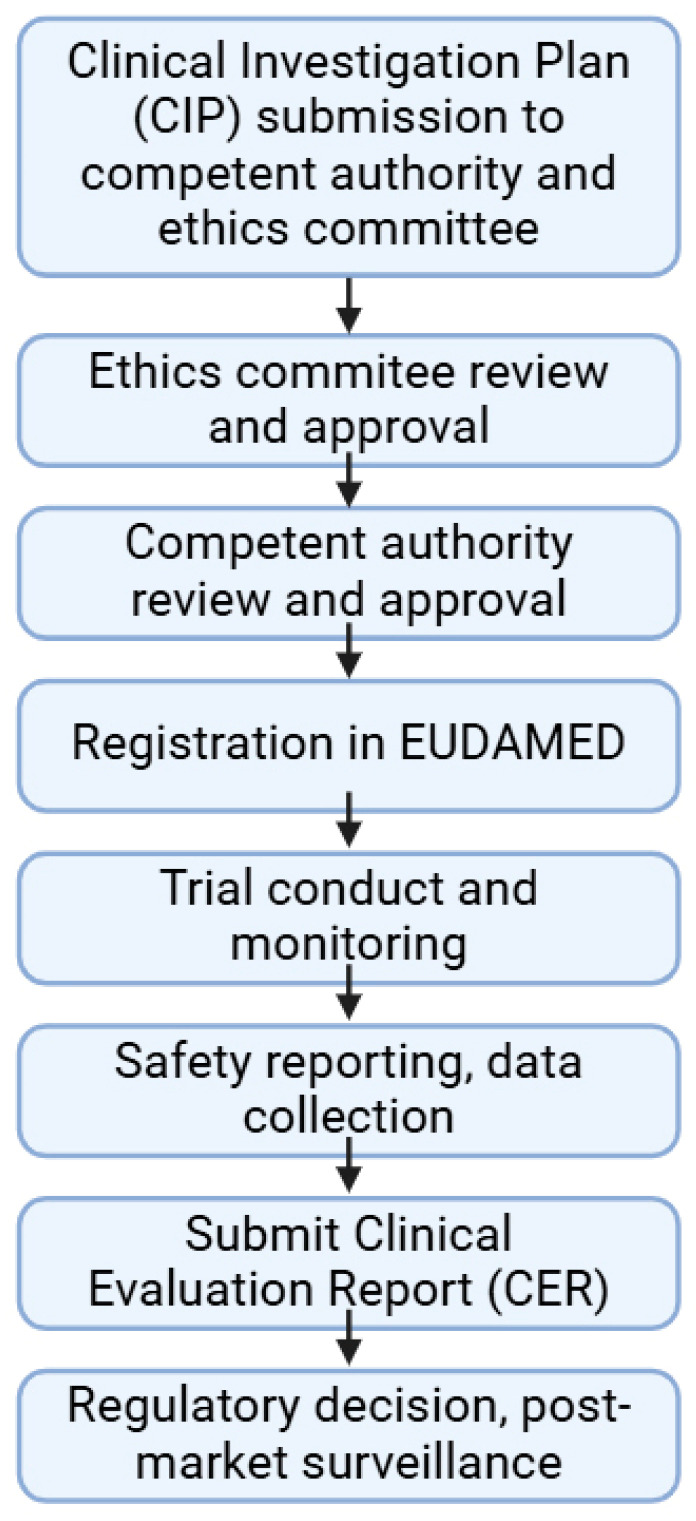
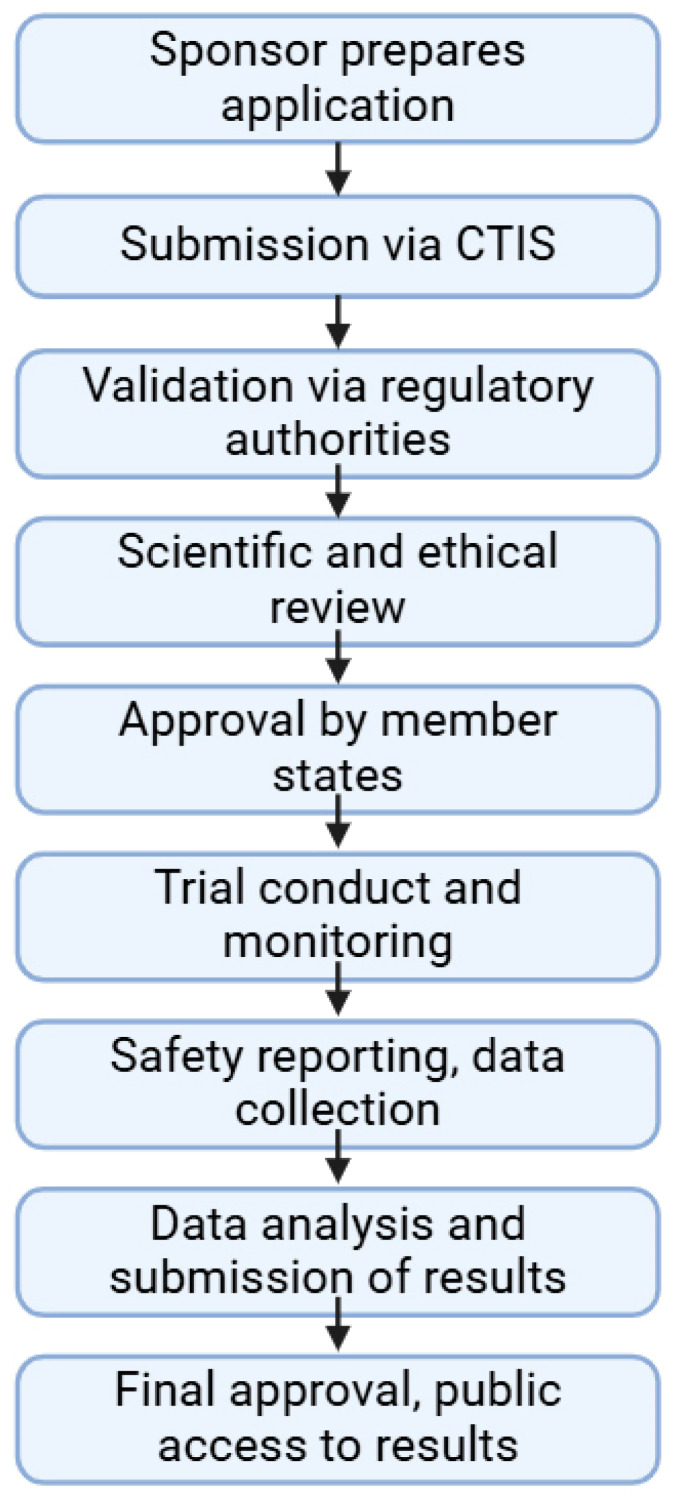
The regulation of clinical trials for medicinal products and medical devices has undergone numerous changes in recent years in the European Union, challenging manufacturers and national regulatory agencies as well. With the introduction of combined drug-device products, the regulatory landscape has been drastically changed to adapt to novel technological advancements and innovations. A comparative analysis has not yet been published highlighting the main differences and common elements of these two medicinal products, which took up almost all of the market in the pharmaceutical sector. Due to stricter regulations in the field of medical devices, the process from application up until post-market surveillance became more difficult, but a correlation between the regulation of drug trials can also be found. The main differences lie in the risk management systems, where, regardless of the background knowledge of a drug, it is always strict and mandatory structured progress, while in the case of medical devices, it is more flexible based on the risk category of the product. Generally, the utilization of e-health opportunities, transparency, and data accessibility have been improved in both fields. Via the adaptation of the mentioned regulation in the EU, the safety of patients and the efficacy of trials have been greatly increased. This manuscript aims to compare the specific regulations of these two types of medicinal products with a brief outlook on the non-EU sector as well.
近年来,欧盟对药品和医疗器械临床试验的监管发生了诸多变化,这对制造商和国家监管机构也构成了挑战。随着联合药物-器械产品的推出,监管格局发生了巨大变化,以适应新的技术进步和创新。尚未有比较分析突出这两种几乎占据了制药行业所有市场的药品的主要差异和共同要素。由于医疗器械领域的法规更加严格,从申请到上市后监测的过程变得更加困难,但药物试验监管之间也存在相关性。主要差异在于风险管理系统,无论药物的背景知识如何,其始终是严格且强制性的结构化流程,而对于医疗器械,基于产品的风险类别则更为灵活。总体而言,两个领域在电子健康机会的利用、透明度和数据可及性方面都有所改善。通过在欧盟采用上述法规,患者安全性和试验有效性都得到了极大提高。本手稿旨在比较这两类药品的具体法规,并简要展望非欧盟领域的情况。