Elder Luke H, Onufriev Alexey V
Department of Computer Science, Virginia Tech, Blacksburg, Virginia 24061, United States.
Department of Physics, Virginia Tech, Blacksburg, Virginia 24061, United States.
J Phys Chem B. 2025 Jul 24;129(29):7483-7498. doi: 10.1021/acs.jpcb.5c02263. Epub 2025 Jul 11.
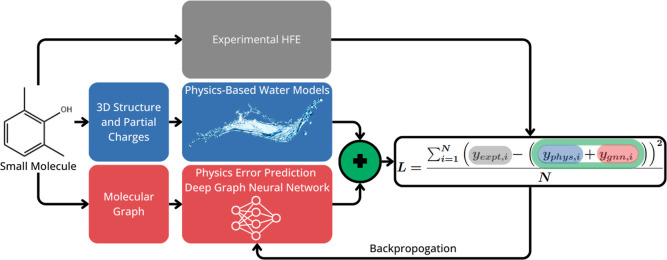
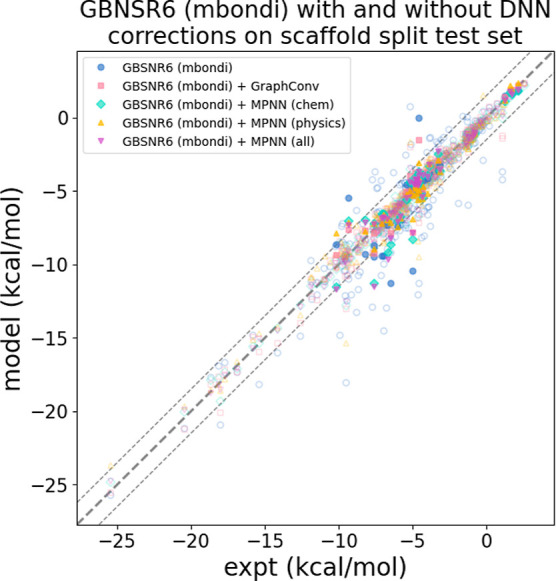
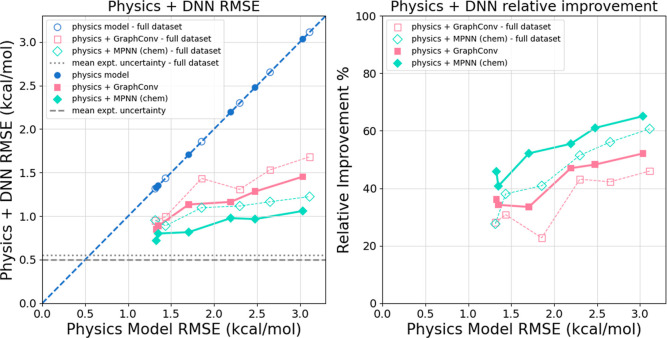
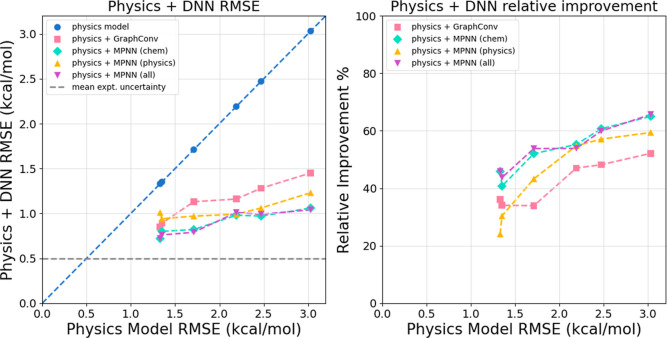
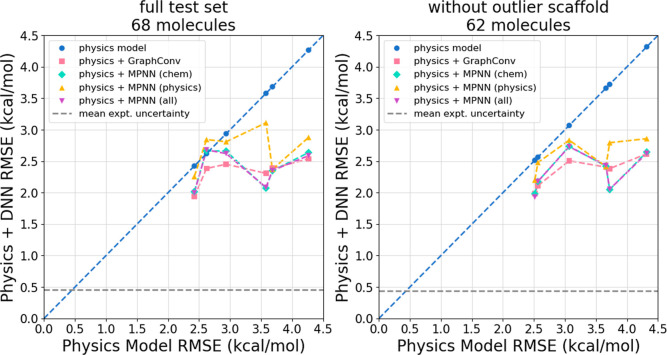
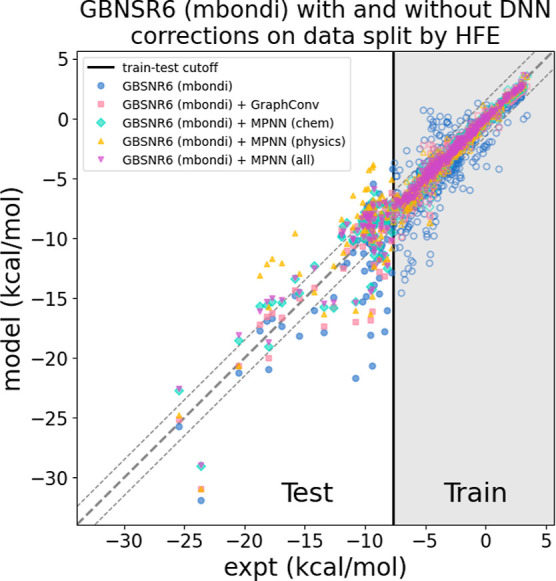
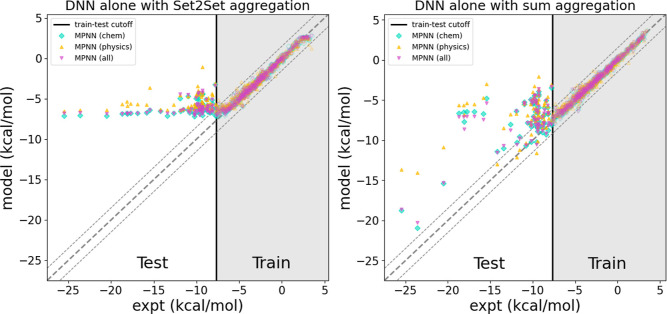
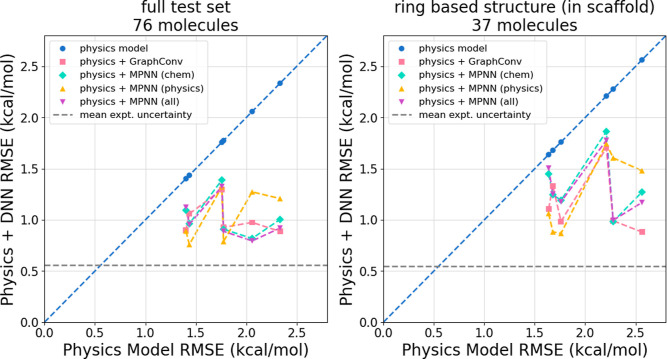
The accuracy of computational water models is crucial to atomistic simulations of biomolecules. Here we explore a decoupled framework that combines classical physics-based models with deep neural networks (DNNs) to correct residual error in hydration free energy (HFE) prediction. Our main goal is to evaluate this framework on out-of-distribution data (molecules that differ significantly from those used in training), where DNNs are known to struggle. Several common physics-based solvation models are used in the evaluation. Graph neural network architectures are tested for their ability to generalize using multiple data set splits, including out-of-distribution HFEs and unseen molecular scaffolds. Our most important finding is that for out-of-distribution data, where DNNs alone often struggle, the physics + DNN models consistently improve physics model predictions. For in-distribution data, the DNN corrections significantly improve the accuracy of physics-based models, with a final RMSE below 1 kcal/mol and a relative improvement between 40% and 65% in most cases. The accuracy of physics + DNN models tends to improve when the 6% of molecules with the highest experimental uncertainty are removed. This study provides insights into the potential and limitations of combining physics and machine learning for molecular modeling, offering a practical and generalizable strategy of using DNN as independent postprocessing correction.
计算水模型的准确性对于生物分子的原子模拟至关重要。在这里,我们探索了一种解耦框架,该框架将基于经典物理的模型与深度神经网络(DNN)相结合,以纠正水合自由能(HFE)预测中的残余误差。我们的主要目标是在分布外数据(与训练中使用的分子有显著差异的分子)上评估这个框架,众所周知,DNN在处理这类数据时会遇到困难。评估中使用了几种常见的基于物理的溶剂化模型。通过多种数据集划分,包括分布外HFE和未见分子支架,测试了图神经网络架构的泛化能力。我们最重要的发现是,对于分布外数据,单独使用DNN往往效果不佳,而物理+DNN模型能持续改进物理模型的预测。对于分布内数据,DNN校正显著提高了基于物理模型的准确性,最终均方根误差(RMSE)低于1千卡/摩尔,在大多数情况下相对提高了40%至65%。去除实验不确定性最高的6%的分子后,物理+DNN模型的准确性往往会提高。这项研究深入探讨了将物理和机器学习结合用于分子建模的潜力和局限性,提供了一种将DNN用作独立后处理校正的实用且可推广的策略。