Bareng Paolo, Wu Kenneth W, Takashima Eizo, Argyropoulos Dionne, Smith Lauren, Naung Myo, Mazhari Ramin, Schoffer Kael, Kiernan-Walker Nicholas, Abraham Anju, Lamont Macie, Mehra Somya, Lim Pailene, Sattabongkot Jetsumon, Monteiro Wuelton, Lacerda Marcus, Healer Julie, Chitnis Chetan E, Tham Wai-Hong, Tsuboi Takafumi, Mueller Ivo, Barry Alyssa E, Longley Rhea J
Centre for Innovation in Infectious Diseases and Immunology Research (CIIDIR), Institute of Mental and Physical Health and Clinical Translation (IMPACT) and School of Medicine, Deakin University, Geelong, VIC, Australia.
Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia.
bioRxiv. 2025 Jul 8:2025.07.07.663616. doi: 10.1101/2025.07.07.663616.
poses a major obstacle to malaria elimination because it can lie dormant in the liver for weeks or months before reactivating and causing a relapse of infection. These dormant forms (hypnozoites) cannot be detected using standard diagnostics, but recent exposure and by proxy, hypnozoite carriage, can be inferred using antibody-based tests (serological markers). In this study, we examined how genetic variation in affects the utility of these antibody markers, and whether redesigned antigens could improve performance.
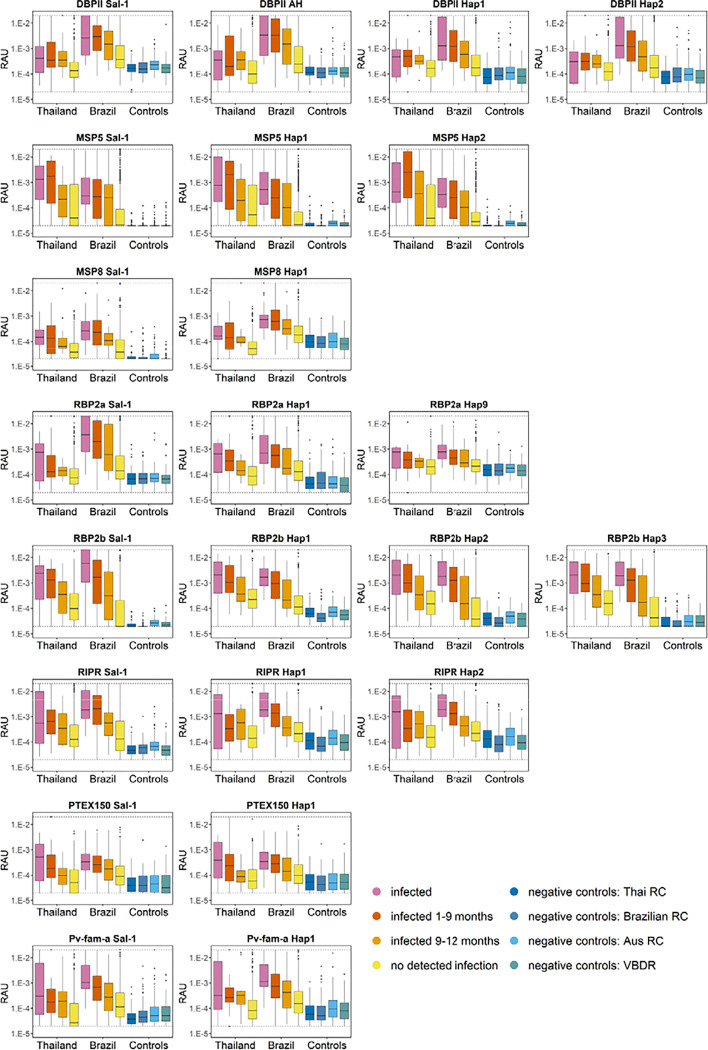
We analysed global genetic data to assess variation in leading serological markers. Based on this, we produced new antigen versions (haplotypes) that better reflect global sequence diversity, compared to the commonly used reference strain (Sal-1). Antibody responses against these new constructs were then tested using samples from well-characterised cohorts in Brazil and Thailand. Antibody levels were assessed in relation to how recently participants had a qPCR-detectable blood-stage infection. We compared the ability of the haplotypes and reference constructs to correctly identify individuals infected within the prior 9-months.
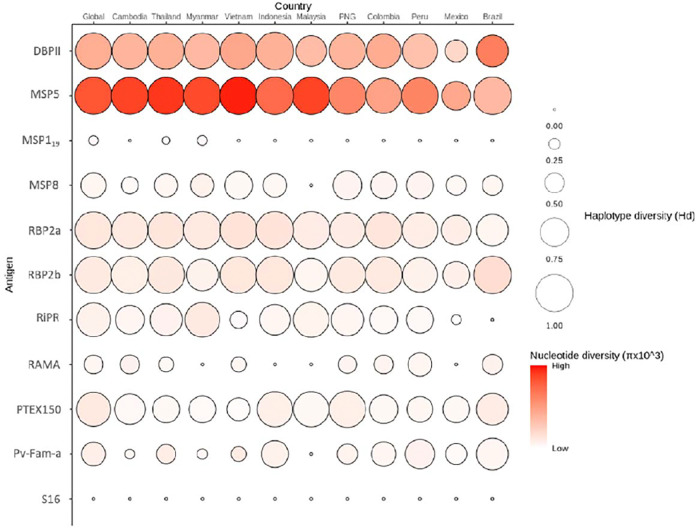
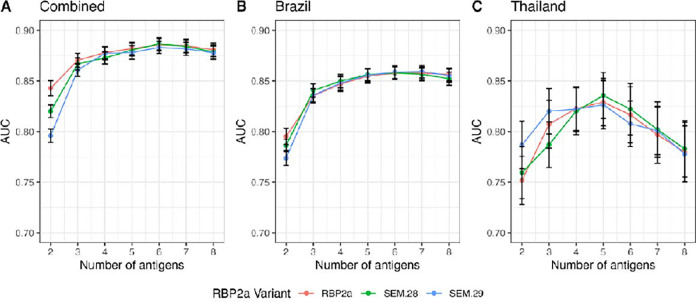
Extensive genetic diversity was identified in two antigens, DBPII and MSP5. Several antigens had large numbers of circulating haplotypes globally, with the percentage with similar sequence identity to the reference Sal-1 ranging from 0.4% (MSP5) to 99% (S16). Two antigens exhibited strong differences in immunogenicity by region and construct (RBP2a and DBPII). However, for most proteins (5 out of 8), these differences had little impact on the accuracy of identifying recent exposure. In cases where performance was affected (e.g. RBP2a), this could be overcome by adding multiple antigens into the classification model.
Even highly diverse antigens can be effective serological exposure markers. Our findings highlight the importance of testing the impact of genetic diversity when designing serological tests and suggest practical strategies, such as using a mix of antigens, to ensure consistent performance across regions.
这对疟疾消除构成了重大障碍,因为它可以在肝脏中潜伏数周或数月,然后重新激活并导致感染复发。这些休眠形式(休眠子)无法通过标准诊断检测到,但近期暴露情况以及由此推断的休眠子携带情况,可以通过基于抗体的检测(血清学标志物)来推断。在本研究中,我们研究了[病原体名称]的基因变异如何影响这些抗体标志物的效用,以及重新设计的抗原是否可以提高检测性能。
我们分析了全球[病原体名称]基因数据,以评估主要血清学标志物的变异情况。在此基础上,我们制备了新的抗原版本(单倍型),与常用的参考菌株(Sal-1)相比,这些新抗原版本能更好地反映全球序列多样性。然后,我们使用来自巴西和泰国特征明确的队列样本,检测了针对这些新构建体的抗体反应。根据参与者最近一次qPCR可检测到的血液阶段[病原体名称]感染时间,评估抗体水平。我们比较了单倍型和参考构建体正确识别在过去9个月内感染个体的能力。
在两种[病原体名称]抗原DBPII和MSP5中发现了广泛的基因多样性。全球有几种抗原存在大量循环单倍型,与参考Sal-1具有相似序列同一性的百分比范围从0.4%(MSP5)到99%(S16)。两种抗原在不同区域和构建体之间表现出强烈的免疫原性差异(RBP2a和DBPII)。然而,对于大多数蛋白质(8种中的5种),这些差异对识别近期暴露的准确性影响不大。在性能受到影响的情况下(如RBP2a),可以通过在分类模型中添加多种抗原加以克服。
即使是高度多样化的抗原也可以成为有效的血清学暴露标志物。我们的研究结果突出了在设计血清学检测时测试基因多样性影响的重要性,并提出了一些实用策略,如使用多种抗原组合,以确保不同区域检测性能的一致性。