Kato Koichi, Asai Haruka, Nakayoshi Tomoki, Mizuno Ayato, Oda Akifumi, Ishikawa Yoshinobu
Faculty of Pharmaceutical Sciences, Shonan University of Medical Sciences, 16-10 Kamishinano, Totsuka-ku, Yokohama 244-0806, Japan.
Faculty of Pharmacy, Meijo University, 150 Yagotoyama, Tempaku-ku, Nagoya 468-8503, Japan.
Int J Mol Sci. 2025 Jul 16;26(14):6819. doi: 10.3390/ijms26146819.
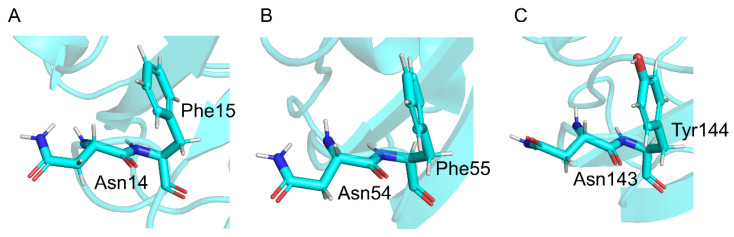
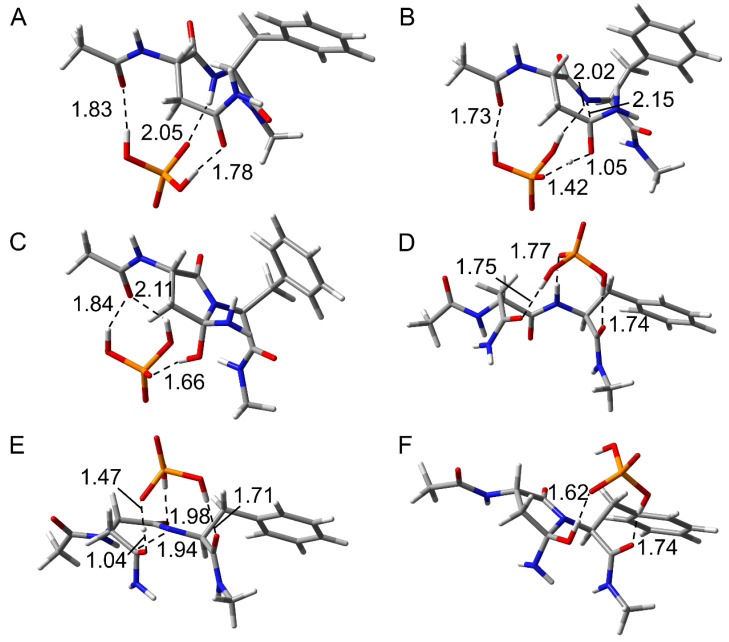
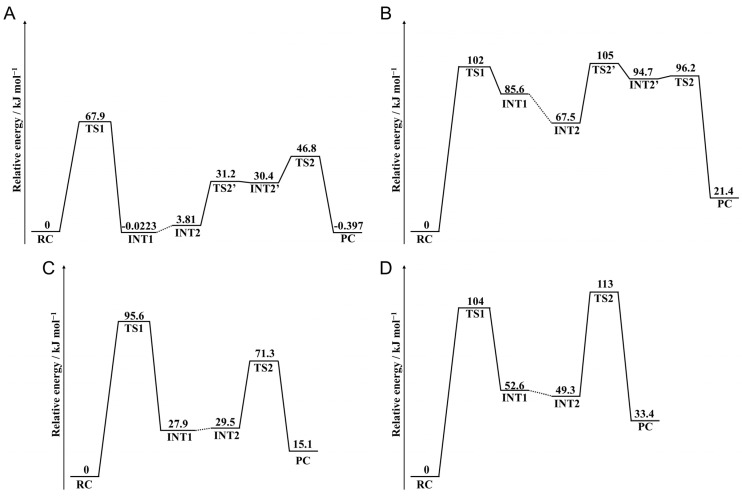
The deamidation rate is relatively high for Asn residues with Phe as the C-terminal adjacent residue in γS-crystallin, which is one of the human crystalline lens proteins. However, peptide-based experiments indicated that bulky amino acid residues on the C-terminal side impaired Asn deamination. In this study, we hypothesized that the side chain of Phe affects the Asn deamidation rate and investigated the succinimide formation process using quantum chemical calculations. The B3LYP density functional theory was used to obtain optimized geometries of energy minima and transition states, and MP2 and M06-2X calculations were used to obtain the single-point energy. Activation barriers and rate-determining step changed depending on the orientation of the Phe side chain. In pathways where an interaction occurred between the benzene ring and the amide group of the Asn residue, the activation barrier was lower than in pathways where this interaction did not occur. Since the aromatic ring is oriented toward the Asn side in experimentally determined structures of γS-crystallin, the above interaction is considered to enhance the Asn deamidation.
在人晶状体蛋白之一的γS-晶状体蛋白中,当Asn残基的C端相邻残基为Phe时,脱酰胺化速率相对较高。然而,基于肽的实验表明,C端侧的大体积氨基酸残基会阻碍Asn脱氨。在本研究中,我们假设Phe的侧链会影响Asn脱酰胺化速率,并使用量子化学计算研究了琥珀酰亚胺的形成过程。采用B3LYP密度泛函理论获得能量最小值和过渡态的优化几何结构,并使用MP2和M06-2X计算获得单点能量。活化能垒和速率决定步骤随Phe侧链的取向而变化。在苯环与Asn残基的酰胺基团之间发生相互作用的途径中,活化能垒低于未发生这种相互作用的途径。由于在实验测定的γS-晶状体蛋白结构中,芳香环朝向Asn侧,因此上述相互作用被认为会增强Asn脱酰胺化。