Sun Xiaoqing, Zhang Xingyu, Li Yuwen, Wen Jiyue, Chen Zhiwu, Chen Shuo
School of Pharmaceutical Sciences, Anhui Medical University, Hefei 230032, China.
The Experimental Research Center, Anhui University of Chinese Medicine, Hefei 230038, China.
Curr Issues Mol Biol. 2025 Jul 3;47(7):513. doi: 10.3390/cimb47070513.
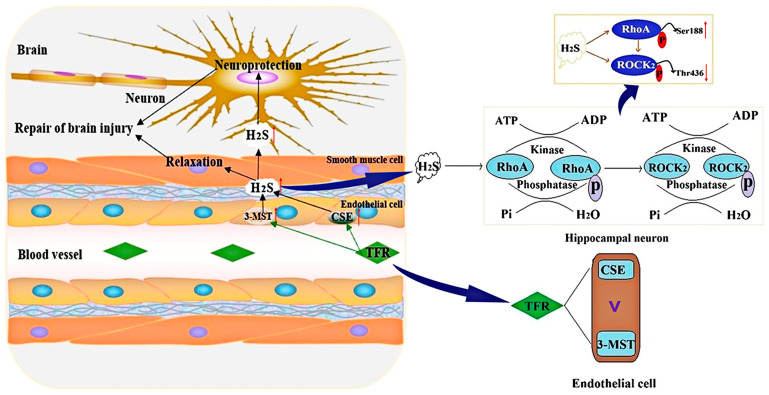
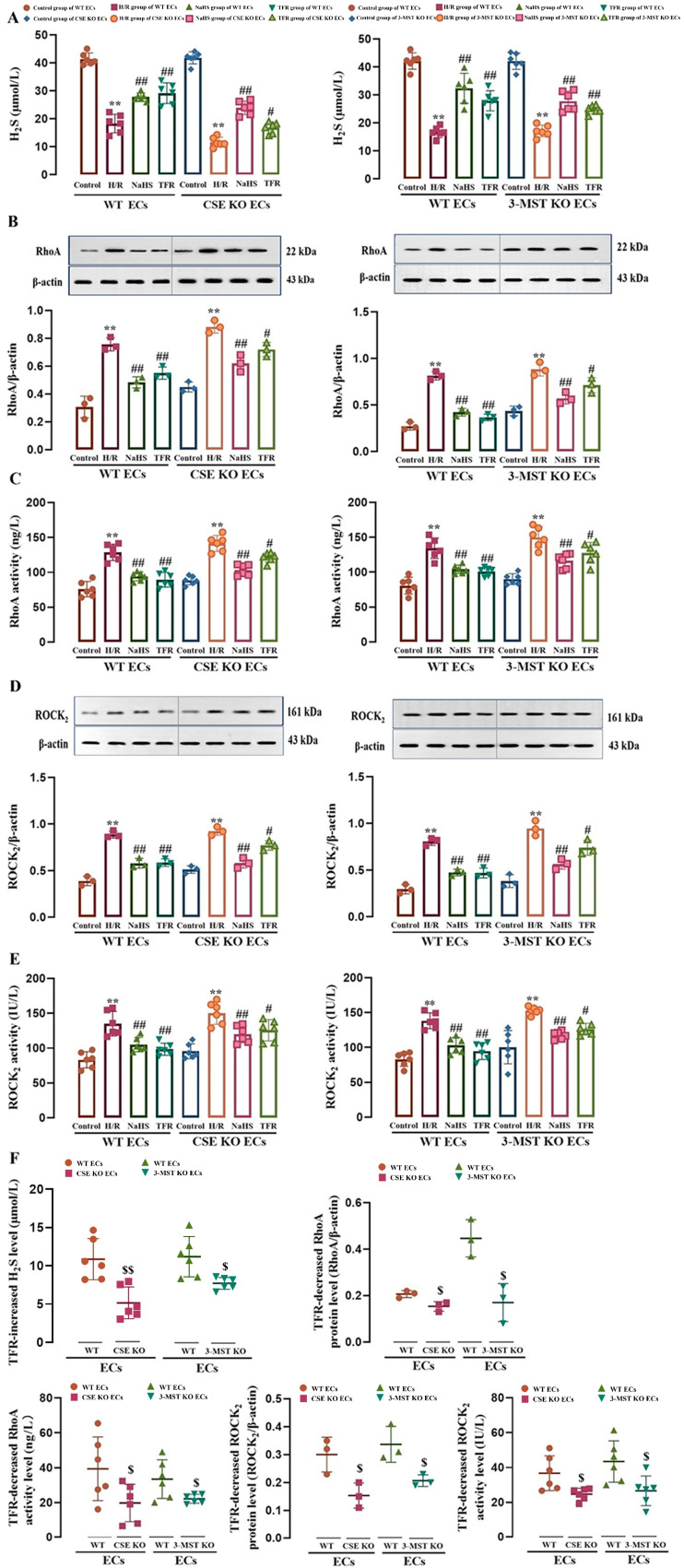
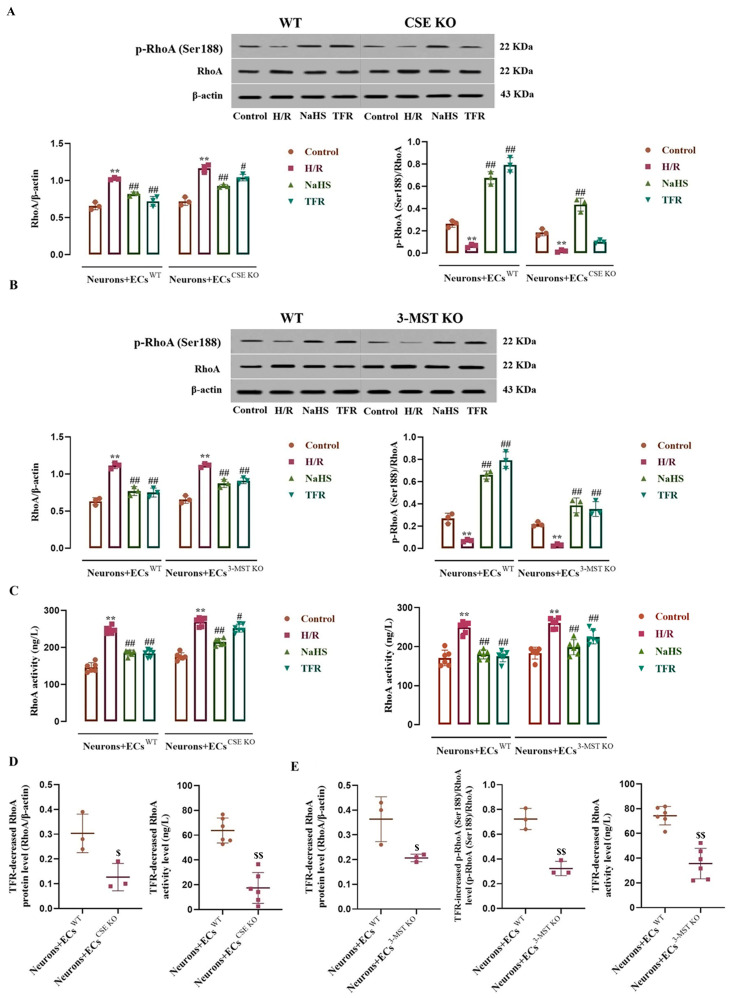
This study aims to investigate the mechanism by which the total flavones of (TFR) protect against cerebral ischemic injury through the endothelial-derived HS-mediated regulation of RhoA phosphorylation at the Ser188 and Rho kinase 2 (ROCK) phosphorylation at Thr436. For experimental design, mouse or rat cerebrovascular endothelial cells (ECs) were cultured with or without neurons and subjected to hypoxia/reoxygenation (H/R) injury. The vasodilation of the cerebral basilar artery was assessed. Cerebral ischemia/reperfusion (I/R) injury was induced in mice by bilateral carotid artery ligation, followed by Morris water maze and open field behavioral assessments. The protein levels of cystathionine-γ-lyase (CSE), 3-mercaptopyruvate sulfurtransferase (3-MST), RhoA, ROCK, p-RhoA (RhoA phosphorylated at Ser188), and p-ROCK (ROCK phosphorylated at Thr436) were quantified. Additionally, the activities of RhoA and ROCK were measured. Notably, TFR significantly inhibited H/R-induced HS reduction and suppressed the increased expression and activity of RhoA and ROCK in ECs, effects attenuated by CSE or 3-MST knockout. Moreover, TFR-mediated cerebrovascular dilation was reduced by RhoA or ROCK inhibitors, while the protective effect of TFR against cerebral I/R injury in mice was markedly attenuated by the heterozygous knockout of ROCK. In the ECs-co-cultured neurons, the inhibition of TFR on H/R-induced neuronal injury and decrease in HS level in the co-culture was attenuated by the knockout of CSE or 3-MST in the ECs. TFR notably inhibited the H/R-induced upregulation of neuronal RhoA, ROCK, and p-ROCK protein levels, as well as the activities of RhoA and ROCK, while reversing the decrease in p-RhoA. However, the knockout of CSE or 3-MST in the ECs significantly attenuated the inhibition of TFR on these increases. Furthermore, 3-MST knockout in ECs attenuated the TFR-mediated suppression of p-RhoA reduction. Additionally, CSE or 3-MST knockout in ECs exacerbated H/R-induced neuronal injury, reduced HS level in the co-culture system, and increased RhoA activity and ROCK expression in neurons. In summary, TFR protected against ischemic cerebral injury by endothelial-derived HS promoting the phosphorylation of RhoA at Ser188 but inhibited the phosphorylation of ROCK at Thr436 to inhibit the RhoA-ROCK pathway in neurons.
本研究旨在探究[具体植物名称]总黄酮(TFR)通过内皮细胞衍生的硫化氢(HS)介导的对丝氨酸188位点RhoA磷酸化及苏氨酸436位点Rho激酶2(ROCK)磷酸化的调控来预防脑缺血损伤的机制。在实验设计中,将小鼠或大鼠脑血管内皮细胞(ECs)在有或无神经元的情况下进行培养,并使其遭受缺氧/复氧(H/R)损伤。评估脑基底动脉的血管舒张情况。通过双侧颈动脉结扎诱导小鼠脑缺血/再灌注(I/R)损伤,随后进行莫里斯水迷宫和旷场行为评估。对胱硫醚-γ-裂解酶(CSE)、3-巯基丙酮酸硫转移酶(3-MST)、RhoA、ROCK、p-RhoA(丝氨酸188位点磷酸化的RhoA)和p-ROCK(苏氨酸436位点磷酸化的ROCK)的蛋白水平进行定量分析。此外,还测量了RhoA和ROCK的活性。值得注意的是,TFR显著抑制H/R诱导的HS减少,并抑制ECs中RhoA和ROCK表达及活性的增加,CSE或3-MST基因敲除可减弱这些作用。此外,RhoA或ROCK抑制剂可降低TFR介导的脑血管舒张,而ROCK杂合基因敲除可显著减弱TFR对小鼠脑I/R损伤的保护作用。在与ECs共培养的神经元中,ECs中CSE或3-MST基因敲除可减弱TFR对H/R诱导的神经元损伤及共培养体系中HS水平降低的抑制作用。TFR显著抑制H/R诱导的神经元RhoA、ROCK和p-ROCK蛋白水平上调以及RhoA和ROCK的活性,同时逆转p-RhoA的降低。然而,ECs中CSE或3-MST基因敲除可显著减弱TFR对这些增加的抑制作用。此外,ECs中3-MST基因敲除可减弱TFR介导的对p-RhoA降低的抑制作用。另外,ECs中CSE或3-MST基因敲除会加剧H/R诱导的神经元损伤,降低共培养体系中的HS水平,并增加神经元中RhoA活性和ROCK表达。总之,TFR通过内皮细胞衍生的HS促进丝氨酸188位点RhoA磷酸化,但抑制苏氨酸436位点ROCK磷酸化来抑制神经元中的RhoA-ROCK途径,从而预防缺血性脑损伤。