Bai Kailun, Moa Belaid, Shao Xiaojian, Zhang Xuekui
Department of Mathematics and Statistics, University of Victoria, Victoria BC, Canada.
Digital Research Alliance of Canada, Victoria BC, Canada.
Comput Struct Biotechnol J. 2025 Jul 23;27:3264-3274. doi: 10.1016/j.csbj.2025.07.019. eCollection 2025.
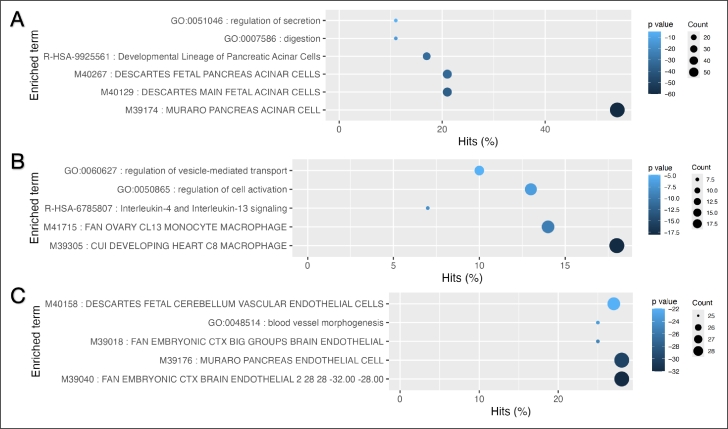
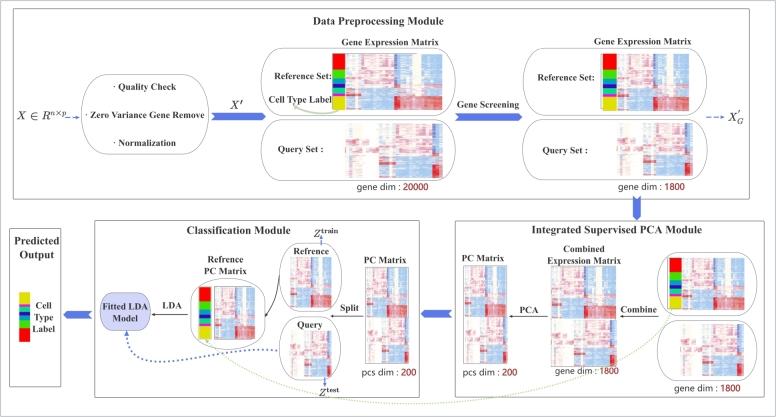
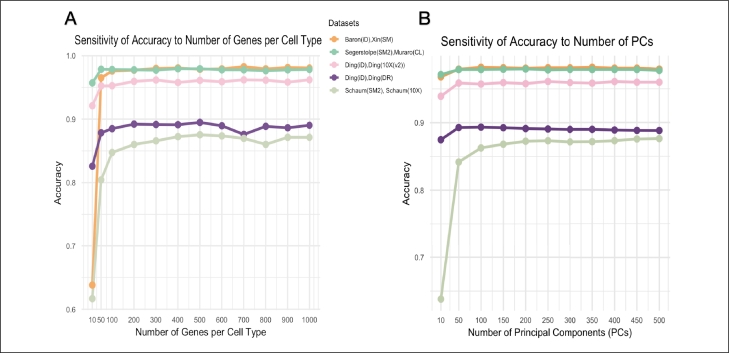
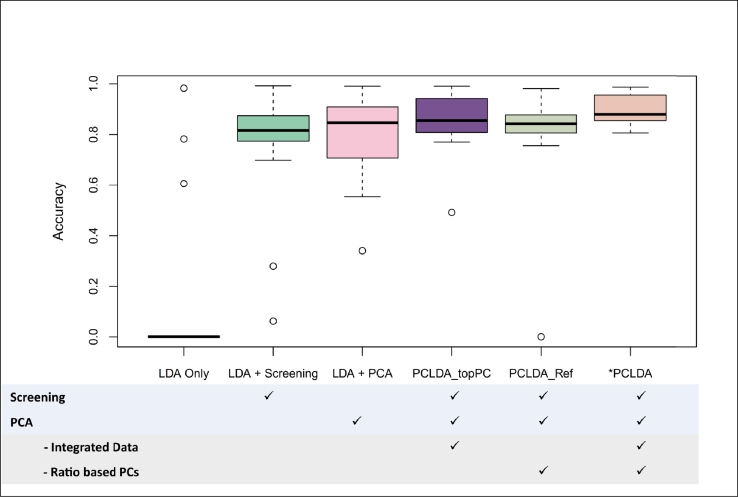
Single-cell RNA sequencing (scRNA-seq) enables high-resolution analysis of cellular heterogeneity, yet accurate and consistent cell-type annotation remains a crucial challenge. Numerous automated tools exist, but their complex modeling assumptions can hinder reliability across varied datasets and protocols. We propose PCLDA, a pipeline composed of three modules: t-test-based gene screening, principal component analysis (PCA) and linear discriminant analysis (LDA), all built on simple statistical methods. An ablation study shows that each module in PCLDA contributes significantly to performance and robustness, with two novel enhancements in the second module yielding substantial gains. Despite these additions, the model retains its original assumptions, computational efficiency, and interpretability. Benchmarking against nine state-of-the-art methods across 22 public scRNA-seq datasets and 35 distinct evaluation scenarios, PCLDA consistently achieves top-tier accuracy under both intra-dataset (cross-validation) and inter-dataset (cross-platform) conditions. Notably, when reference and query data are generated via different protocols, PCLDA remains stable and often outperforms more complex machine-learning approaches. Furthermore, PCLDA offers strong interpretability, attributed to the linear nature of its PCA and LDA modules. The final decision boundaries are linear combinations of the original gene expression values, directly reflecting the contribution of each gene to the classification. Top-weighted genes identified by PCLDA better capture biologically meaningful signals in enrichment analyses than those selected via marginal screening alone, offering deeper functional insights into cell-type specificity. In conclusion, our work underscores the utility of carefully enhanced simple statistics methods for single-cell annotation. PCLDA's simplicity, interpretability, and consistently high performance make it a practical, reliable alternative to more complex annotation pipelines. Code is available on GitHub:https://github.com/kellen8hao/PCLDA.
单细胞RNA测序(scRNA-seq)能够对细胞异质性进行高分辨率分析,但准确且一致的细胞类型注释仍然是一项关键挑战。虽然存在许多自动化工具,但其复杂的建模假设可能会妨碍在不同数据集和实验方案中的可靠性。我们提出了PCLDA,这是一个由三个模块组成的流程:基于t检验的基因筛选、主成分分析(PCA)和线性判别分析(LDA),所有这些都建立在简单的统计方法之上。一项消融研究表明,PCLDA中的每个模块对性能和稳健性都有显著贡献,第二个模块中的两项新颖改进带来了显著提升。尽管有这些改进,该模型仍保留其原始假设、计算效率和可解释性。在22个公共scRNA-seq数据集和35种不同评估场景下与九种先进方法进行基准测试时,PCLDA在数据集内(交叉验证)和数据集间(跨平台)条件下均始终实现顶级准确性。值得注意的是,当参考数据和查询数据通过不同实验方案生成时,PCLDA保持稳定,并且通常优于更复杂的机器学习方法。此外,由于其PCA和LDA模块的线性性质,PCLDA具有很强的可解释性。最终的决策边界是原始基因表达值的线性组合,直接反映了每个基因对分类的贡献。与仅通过边际筛选选择的基因相比,PCLDA识别出的权重最高的基因在富集分析中能更好地捕捉生物学上有意义的信号,从而为细胞类型特异性提供更深入的功能见解。总之,我们的工作强调了精心改进的简单统计方法在单细胞注释中的实用性。PCLDA的简单性、可解释性和始终如一的高性能使其成为更复杂注释流程的实用、可靠替代方案。代码可在GitHub上获取:https://github.com/kellen8hao/PCLDA。