Center for Computational Biology, University of California, Berkeley, CA, USA.
Department of Electrical Engineering and Computer Sciences, University of California, Berkeley, CA, USA.
Mol Syst Biol. 2021 Jan;17(1):e9620. doi: 10.15252/msb.20209620.
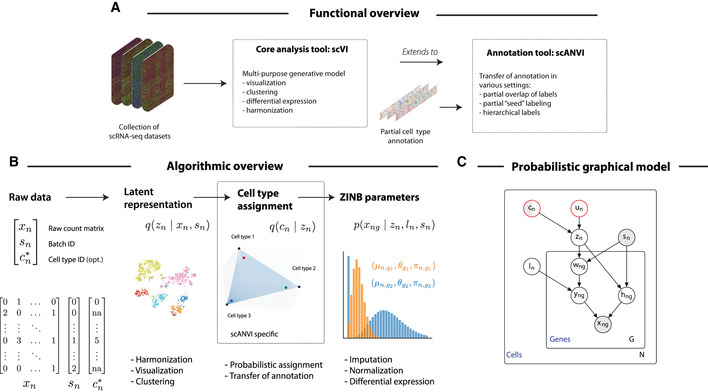
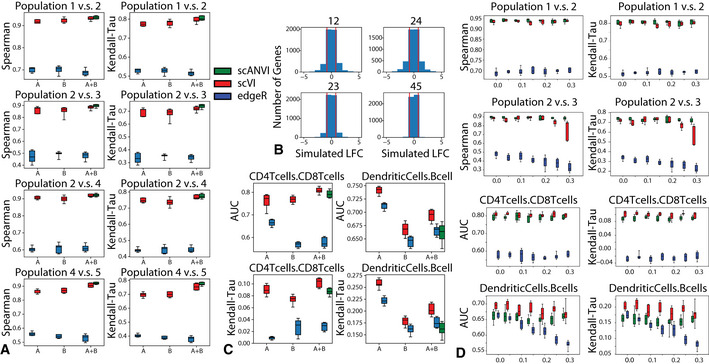
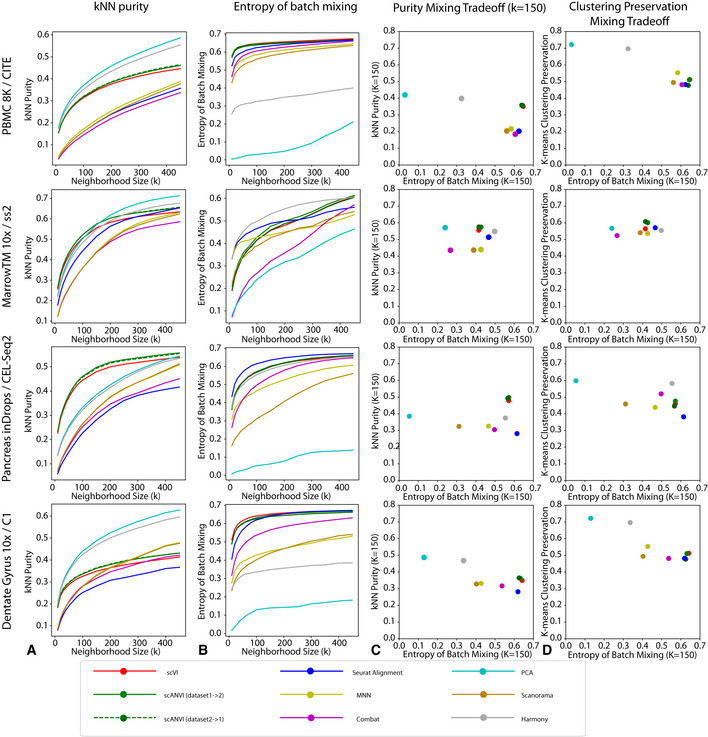
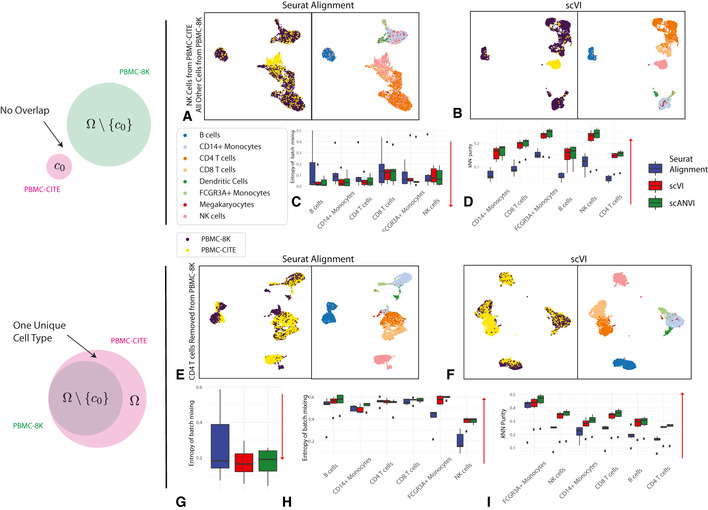
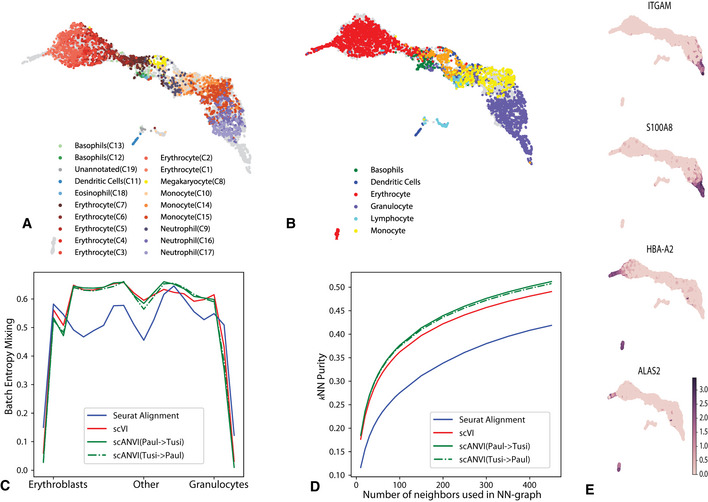
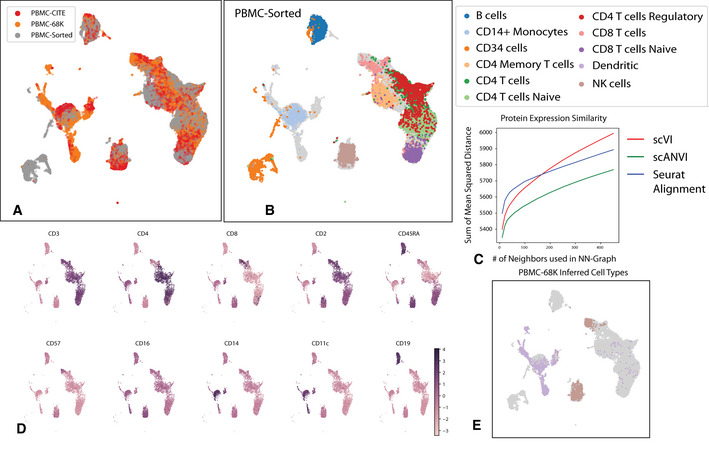
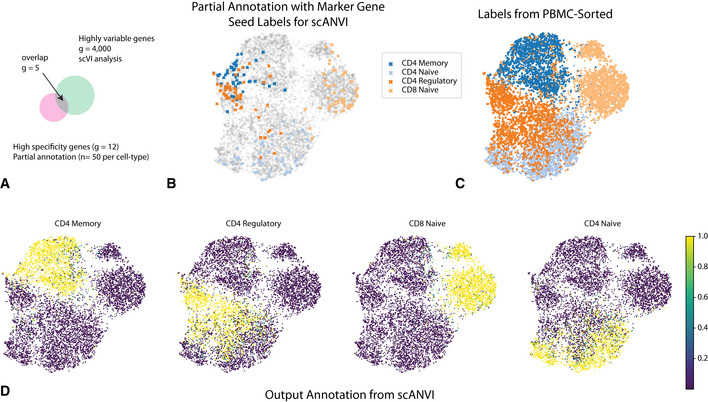
As the number of single-cell transcriptomics datasets grows, the natural next step is to integrate the accumulating data to achieve a common ontology of cell types and states. However, it is not straightforward to compare gene expression levels across datasets and to automatically assign cell type labels in a new dataset based on existing annotations. In this manuscript, we demonstrate that our previously developed method, scVI, provides an effective and fully probabilistic approach for joint representation and analysis of scRNA-seq data, while accounting for uncertainty caused by biological and measurement noise. We also introduce single-cell ANnotation using Variational Inference (scANVI), a semi-supervised variant of scVI designed to leverage existing cell state annotations. We demonstrate that scVI and scANVI compare favorably to state-of-the-art methods for data integration and cell state annotation in terms of accuracy, scalability, and adaptability to challenging settings. In contrast to existing methods, scVI and scANVI integrate multiple datasets with a single generative model that can be directly used for downstream tasks, such as differential expression. Both methods are easily accessible through scvi-tools.
随着单细胞转录组学数据集数量的增加,下一步自然是整合积累的数据,以实现细胞类型和状态的通用本体。然而,直接比较数据集之间的基因表达水平,并根据现有注释在新数据集中自动分配细胞类型标签并不容易。在本文中,我们证明了我们之前开发的方法 scVI 为联合表示和分析 scRNA-seq 数据提供了一种有效且完全概率的方法,同时考虑了由生物和测量噪声引起的不确定性。我们还介绍了使用变分推理进行单细胞注释 (scANVI),这是 scVI 的半监督变体,旨在利用现有的细胞状态注释。我们证明,scVI 和 scANVI 在数据集成和细胞状态注释方面的准确性、可扩展性以及对挑战性设置的适应性方面均优于最先进的方法。与现有方法不同,scVI 和 scANVI 可以使用单个生成模型整合多个数据集,该模型可以直接用于下游任务,如差异表达。这两种方法都可以通过 scvi-tools 轻松访问。