Kim So Young, Lee Bin, Lee Je-Jung, Kwak Man Sup, Rhee Woo Joong, Park In Ho, Shin Jeon-Soo
Department of Microbiology, Yonsei University College of Medicine, Seoul, South Korea.
Brain Korea 21 FOUR Project for Medical Science, Yonsei University College of Medicine, Seoul, South Korea.
Cell Death Discov. 2025 Aug 29;11(1):416. doi: 10.1038/s41420-025-02708-1.
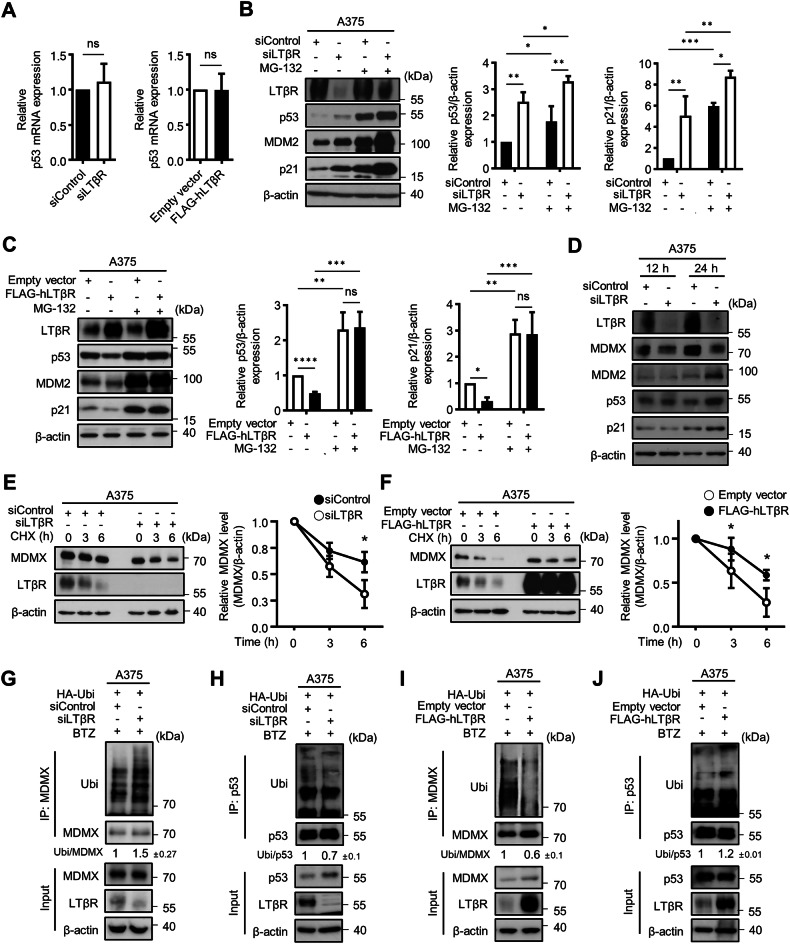
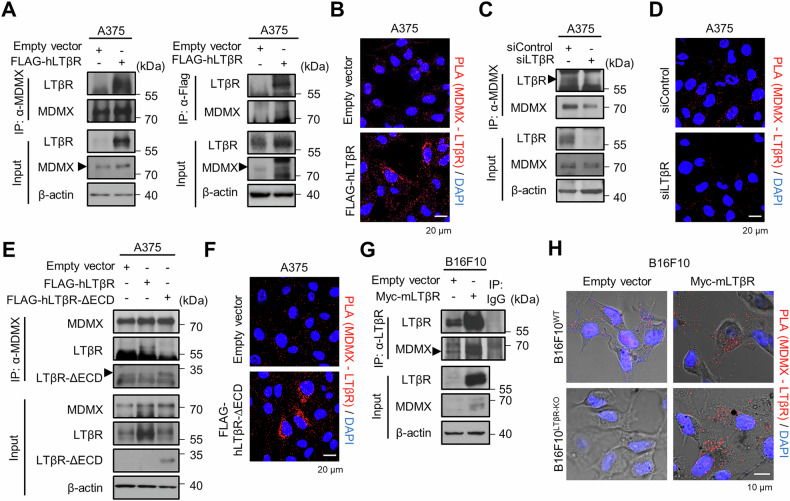
The lymphotoxin β receptor (LTβR), a key activator of non-canonical NF-κB signaling, is expressed in various cells, including cancer cells. Although high expression of LTβR has been associated with poor patient prognosis and drug resistance, conflicting evidence suggested that LTβR induces apoptosis. To investigate the functional role of LTβR in tumors, we performed LTβR knockdown in cancer cells. We found that LTβR knockdown induced senescence phenomena such as reduced cell number; increased cell size; increased SA-β-Gal activity; and upregulated p53, MDM2 and p21 expression. Moreover, LTβR knockdown induced p21-mediated senescence in p53 WT cancer cells, but not in p53 mutant cancer cells. The level of p53 is regulated by MDM2 and MDMX; MDMX enhances MDM2 activity but is also subject to MDM2-mediated degradation in the nucleus. We found that the intracellular domain of LTβR bound to MDMX thereby inhibited its nuclear translocation, which in turn reduced MDMX ubiquitination and consequently promoted p53 ubiquitination. Additionally, tumors derived from B16F10 cells in WT mice exhibited significantly reduced growth compared to those derived from B16F10 cells. These results demonstrate that LTβR regulates p53 protein levels by modulating MDMX stability and localization, resulting in p53-mediated cellular senescence. LTβR regulates p53-mediated senescence by inhibiting MDMX nuclear translocation and degradation. LTβR interacts with MDMX in the cytoplasm, preventing its nuclear translocation and degradation under normal conditions (dotted arrows). When LTβR is depleted, MDMX is translocated into the nucleus by MDM2, and undergoes degradation (solid arrows). This reduces p53 degradation and consequently activates p53, leading to p21 transcription and the induction of cellular senescence. Treatment with doxorubicin (Dox) or nutlin-3a further enhances p53-mediated transcriptional activation of p21, and their combination with LTβR depletion exerts an additive effect in promoting cellular senescence.
淋巴毒素β受体(LTβR)是非经典NF-κB信号通路的关键激活因子,在包括癌细胞在内的多种细胞中表达。尽管LTβR的高表达与患者预后不良和耐药性相关,但有相互矛盾的证据表明LTβR可诱导细胞凋亡。为了研究LTβR在肿瘤中的功能作用,我们在癌细胞中进行了LTβR基因敲低。我们发现,LTβR基因敲低诱导了衰老现象,如细胞数量减少、细胞大小增加、SA-β-Gal活性增加以及p53、MDM2和p21表达上调。此外,LTβR基因敲低在p53野生型癌细胞中诱导了p21介导的衰老,但在p53突变型癌细胞中未诱导。p53的水平受MDM2和MDMX调节;MDMX增强MDM2活性,但在细胞核中也会受到MDM2介导的降解。我们发现,LTβR的细胞内结构域与MDMX结合,从而抑制其核转位,进而减少MDMX泛素化,从而促进p53泛素化。此外,与源自B16F10细胞的肿瘤相比,野生型小鼠中源自B16F10细胞的肿瘤生长显著降低。这些结果表明,LTβR通过调节MDMX的稳定性和定位来调节p53蛋白水平,从而导致p53介导的细胞衰老。LTβR通过抑制MDMX核转位和降解来调节p53介导的衰老。LTβR在细胞质中与MDMX相互作用,在正常条件下阻止其核转位和降解(虚线箭头)。当LTβR缺失时,MDMX通过MDM2转运到细胞核中并发生降解(实线箭头)。这减少了p53降解,从而激活p53,导致p21转录并诱导细胞衰老。用阿霉素(Dox)或nutlin-3a处理进一步增强了p53介导的p21转录激活,它们与LTβR缺失联合使用在促进细胞衰老方面发挥了相加作用。