Brouillet E, Hantraye P, Ferrante R J, Dolan R, Leroy-Willig A, Kowall N W, Beal M F
Departement de Recherche en Imagerie, Pharmacologie, et Physiologie, Commissariat à la Energie Atomique-Direction des Sciences du Vivant, Orsay, France.
Proc Natl Acad Sci U S A. 1995 Jul 18;92(15):7105-9. doi: 10.1073/pnas.92.15.7105.
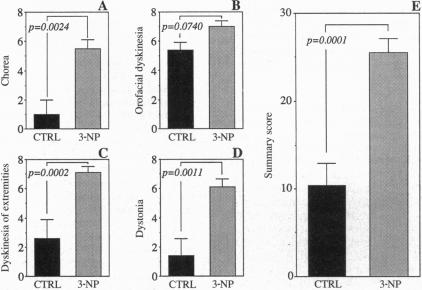
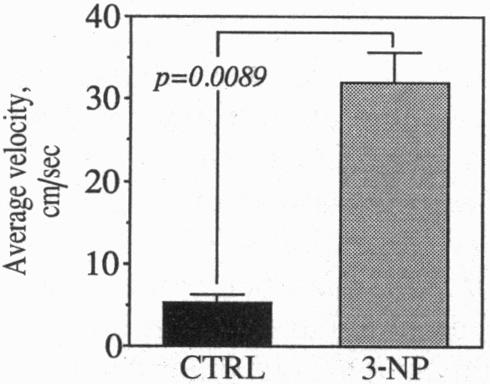
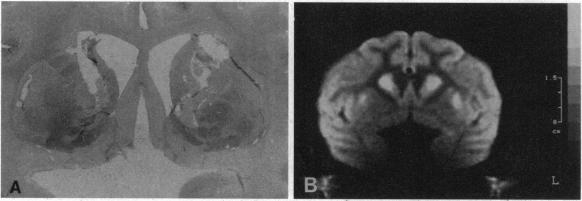
Although the gene defect responsible for Huntington disease (HD) has recently been identified, the pathogenesis of the disease remains obscure. One potential mechanism is that the gene defect may lead to an impairment of energy metabolism followed by slow excitotoxic neuronal injury. In the present study we examined whether chronic administration of 3-nitropropionic acid (3-NP), an irreversible inhibitor of succinate dehydrogenase, can replicate the neuropathologic and clinical features of HD in nonhuman primates. After 3-6 weeks of 3-NP administration, apomorphine treatment induced a significant increase in motor activity as compared with saline-treated controls. Animals showed both choreiform movements, as well as foot and limb dystonia, which are characteristic of HD. More prolonged 3-NP treatment in two additional primates resulted in spontaneous dystonia and dyskinesia accompanied by lesions in the caudate and putamen seen by magnetic resonance imaging. Histologic evaluation showed that there was a depletion of calbindin neurons, astrogliosis, sparing of NADPH-diaphorase neurons, and growth-related proliferative changes in dendrites of spiny neurons similar to changes in HD. The striosomal organization of the striatum and the nucleus accumbens were spared. These findings show that chronic administration of 3-NP to nonhuman primates can replicate many of the characteristic motor and histologic features of HD, further strengthening the possibility that a subtle impairment of energy metabolism may play a role in its pathogenesis.
尽管导致亨廷顿病(HD)的基因缺陷最近已被确定,但该病的发病机制仍不清楚。一种潜在机制是,基因缺陷可能导致能量代谢受损,进而引发缓慢的兴奋性毒性神经元损伤。在本研究中,我们检测了长期给予琥珀酸脱氢酶的不可逆抑制剂3-硝基丙酸(3-NP)是否能在非人灵长类动物中重现HD的神经病理学和临床特征。给予3-NP 3至6周后,与给予生理盐水的对照组相比,阿扑吗啡治疗使运动活性显著增加。动物表现出舞蹈样动作以及足部和肢体肌张力障碍,这些都是HD的特征。在另外两只灵长类动物中延长3-NP治疗时间,导致出现自发性肌张力障碍和运动障碍,同时磁共振成像显示尾状核和壳核有病变。组织学评估显示,钙结合蛋白神经元减少、星形胶质细胞增生、NADPH-黄递酶神经元未受影响,并且棘状神经元树突出现与HD变化相似的生长相关增殖性改变。纹状体和伏隔核的纹状小体组织未受影响。这些发现表明,长期给非人灵长类动物施用3-NP可以重现HD的许多典型运动和组织学特征,进一步增强了能量代谢的细微损害可能在其发病机制中起作用的可能性。