Liu Huajun, Atkins Joshua, Kass Robert S
Department of Pharmacology, College of Physicians and Surgeons of Columbia University, New York, NY 10032, USA.
J Gen Physiol. 2003 Mar;121(3):199-214. doi: 10.1085/jgp.20028723.
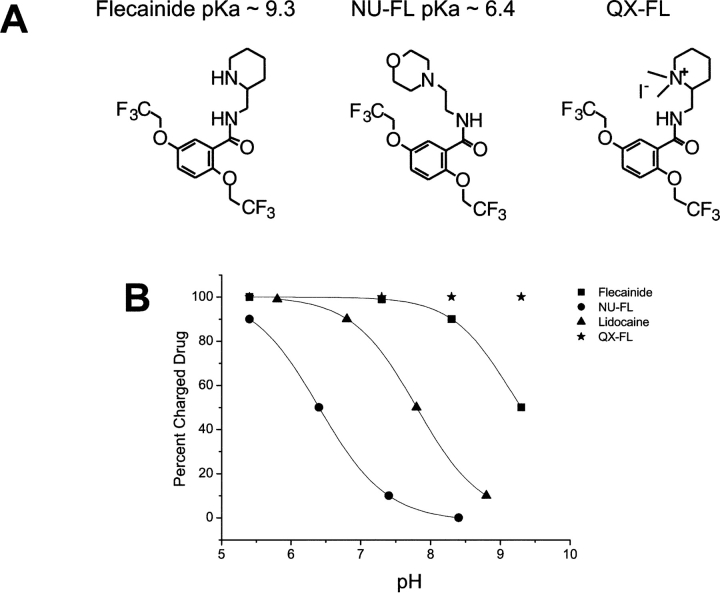
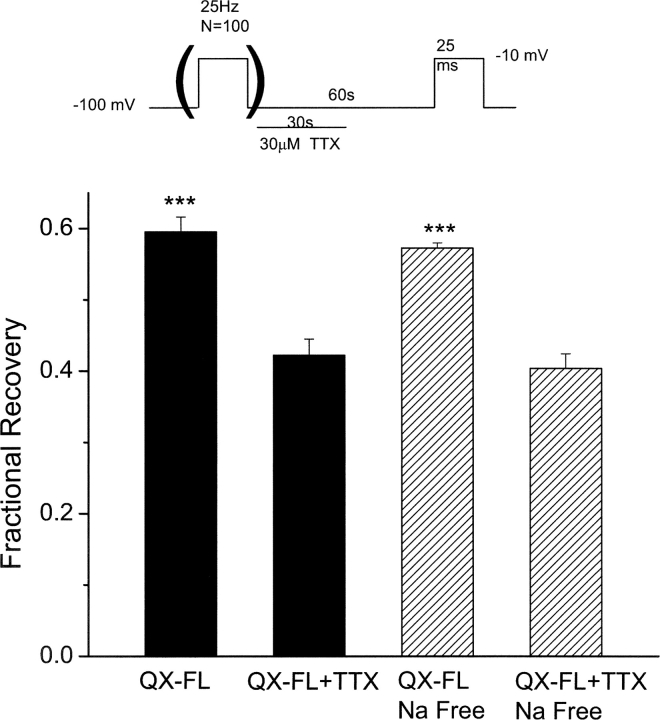
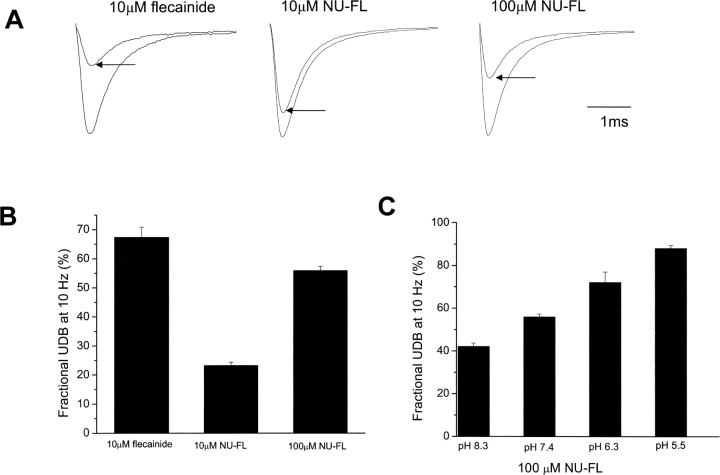
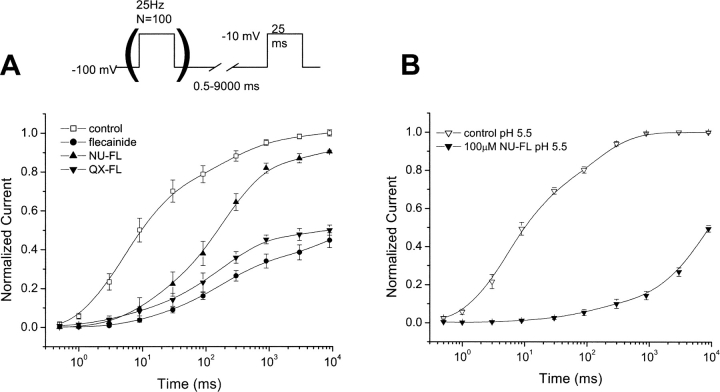
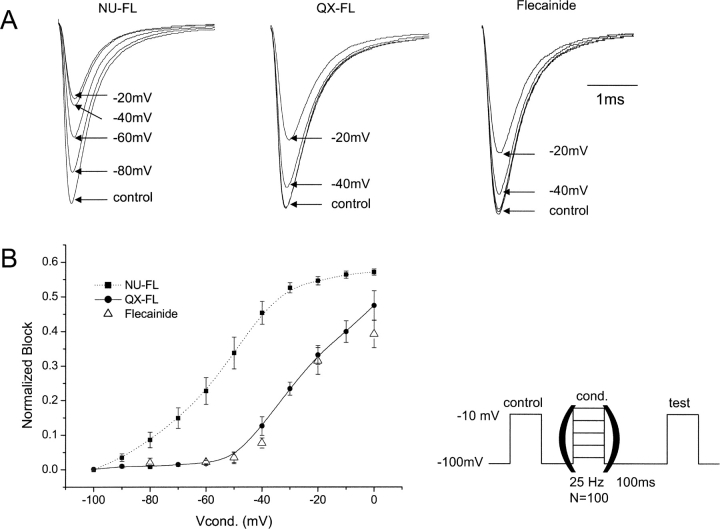
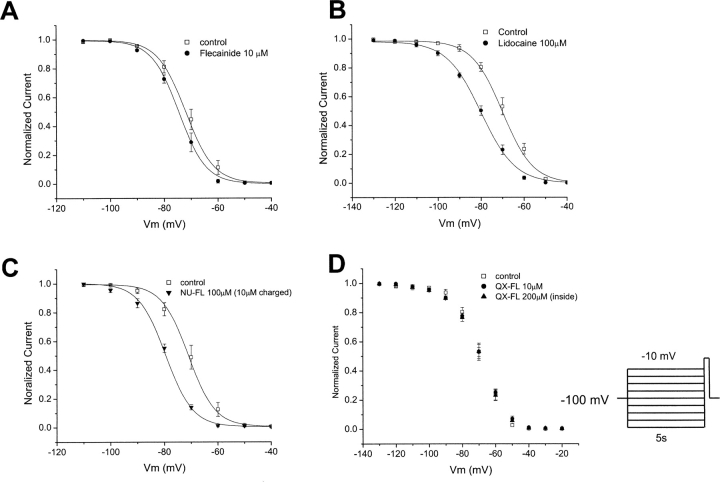
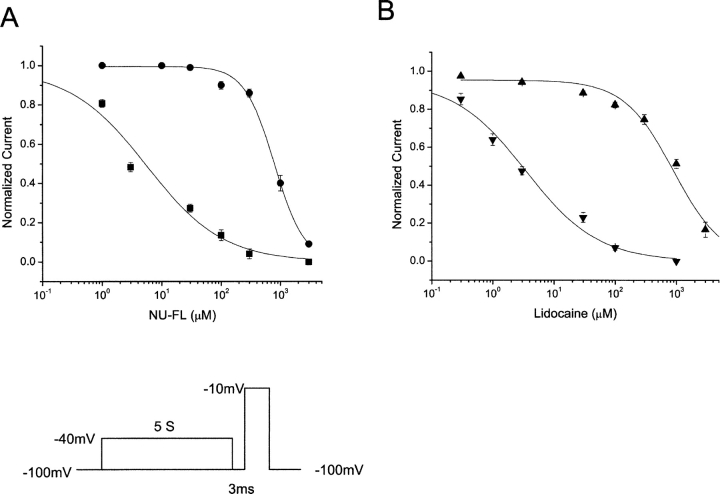
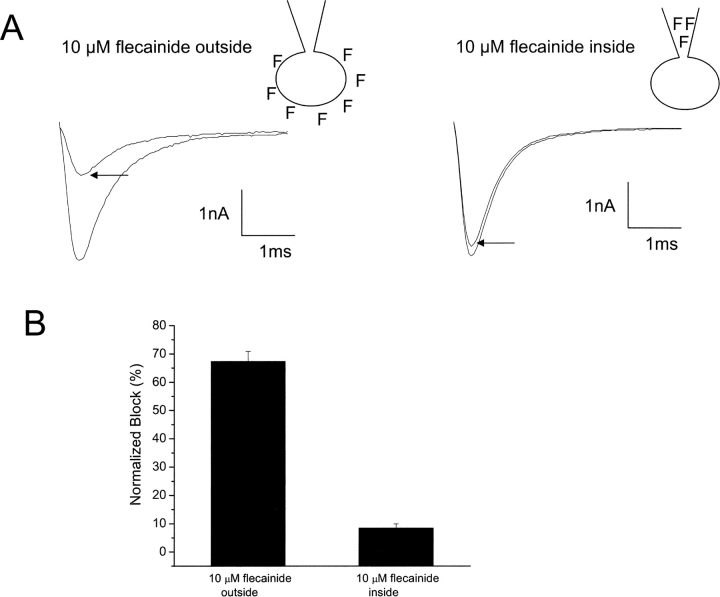
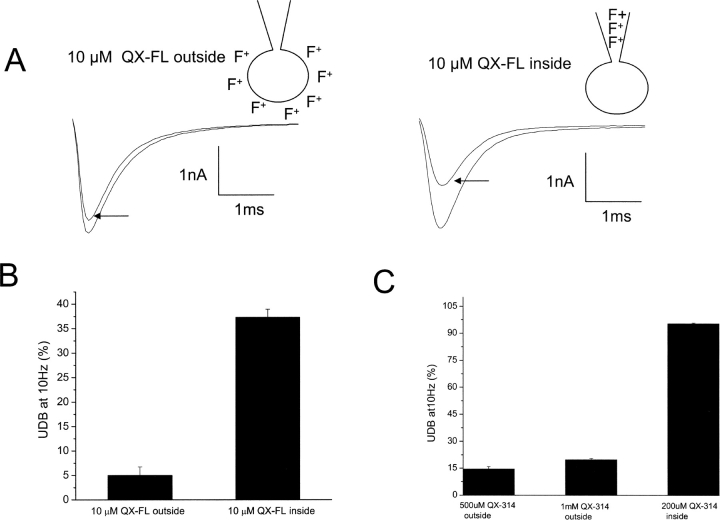
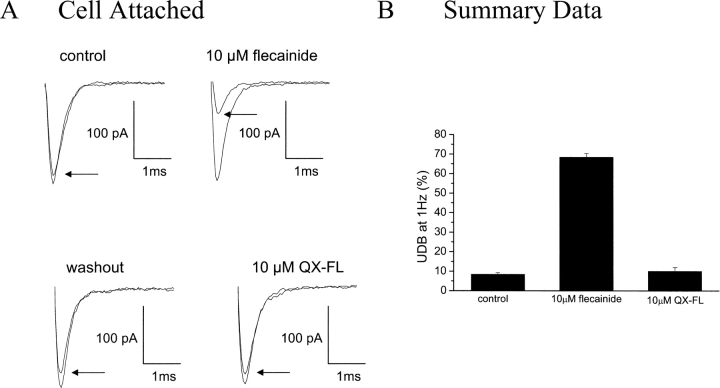
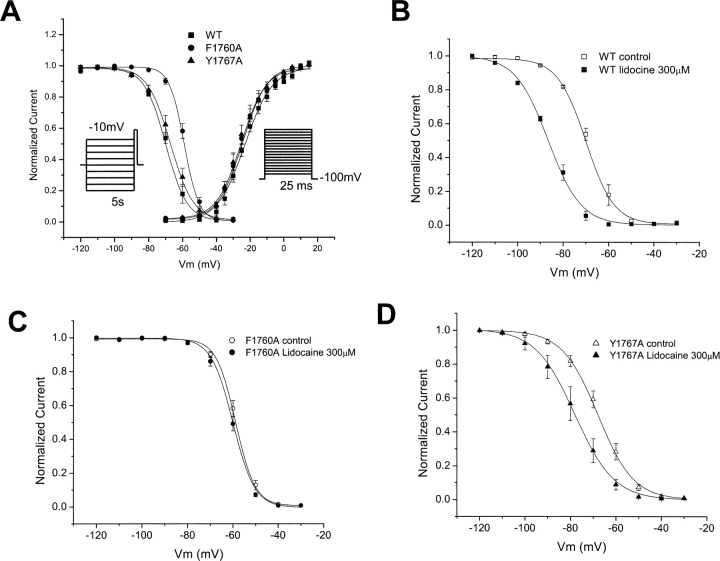
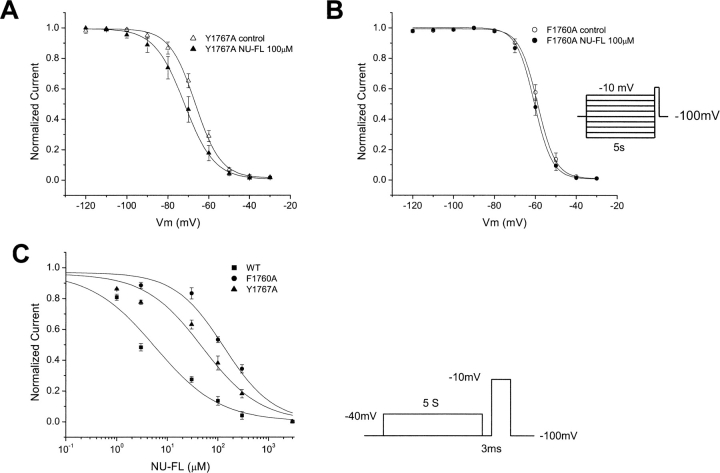
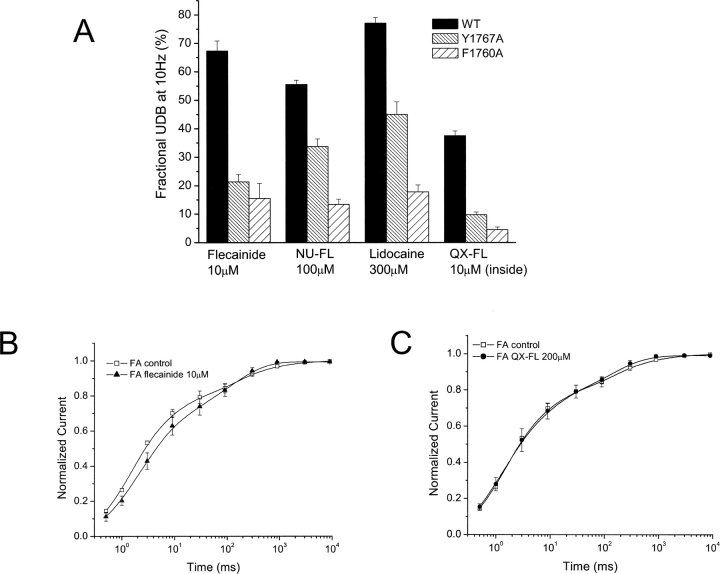
Flecainide (pKa 9.3, 99% charged at pH 7.4) and lidocaine (pKa 7.6-8.0, approximately 50% neutral at pH 7.4) have similar structures but markedly different effects on Na(+) channel activity. Both drugs cause well-characterized use-dependent block (UDB) of Na(+) channels due to stabilization of the inactivated state, but flecainide requires that channels first open before block develops, whereas lidocaine is believed to bind directly to the inactivated state. To test whether the charge on flecainide might determine its state specificity of Na(+) channel blockade, we developed two flecainide analogues, NU-FL (pKa 6.4), that is 90% neutral at pH 7.4, and a quaternary flecainide analogue, QX-FL, that is fully charged at physiological pH. We examined the effects of flecainide, NU-FL, QX-FL, and lidocaine on human cardiac Na(+) channels expressed in human embryonic kidney (HEK) 293 cells. At physiological pH, NU-FL, like lidocaine but not flecainide, interacts preferentially with inactivated channels without prerequisite channel opening, and causes minimal UDB. We find that UDB develops predominantly by the charged form of flecainide as evidenced by investigation of QX-FL at physiological pH and NU-FL investigated over a more acidic pH range where its charged fraction is increased. QX-FL is a potent blocker of channels when applied from inside the cell, but acts very weakly with external application. UDB by QX-FL, like flecainide, develops only after channels open. Once blocked, channels recover very slowly from QX-FL block, apparently without requisite channel opening. Our data strongly suggest that it is the difference in degree of ionization (pKa) between lidocaine and flecainide, rather than gross structural features, that determines distinction in block of cardiac Na(+) channels. The data also suggest that the two drugs share a common receptor but, consistent with the modulated receptor hypothesis, reach this receptor by distinct routes dictated by the degree of ionization of the drug molecules.
氟卡尼(pKa 9.3,在pH 7.4时99%呈离子化)和利多卡因(pKa 7.6 - 8.0,在pH 7.4时约50%呈中性)结构相似,但对Na⁺通道活性的影响却显著不同。由于失活状态的稳定,两种药物都会导致特征明确的Na⁺通道使用依赖性阻滞(UDB),但氟卡尼需要通道先开放才会产生阻滞,而利多卡因被认为是直接与失活状态结合。为了测试氟卡尼上的电荷是否可能决定其对Na⁺通道阻滞的状态特异性,我们开发了两种氟卡尼类似物,NU - FL(pKa 6.4),在pH 7.4时90%呈中性,以及一种季铵化氟卡尼类似物QX - FL,在生理pH下完全呈离子化。我们研究了氟卡尼、NU - FL、QX - FL和利多卡因对在人胚肾(HEK)293细胞中表达的人心脏Na⁺通道的影响。在生理pH下,NU - FL与利多卡因一样,而不像氟卡尼,优先与失活通道相互作用,无需通道预先开放,并且产生的UDB最小。我们发现,如在生理pH下对QX - FL以及在更酸性pH范围内对NU - FL(其离子化部分增加)进行研究所证明的,UDB主要由氟卡尼的离子化形式产生。当从细胞内施加时,QX - FL是通道的强效阻滞剂,但外用时作用非常弱。与氟卡尼一样,QX - FL引起的UDB仅在通道开放后才出现。一旦被阻滞,通道从QX - FL阻滞中恢复非常缓慢,显然无需通道开放。我们的数据强烈表明,决定心脏Na⁺通道阻滞差异的是利多卡因和氟卡尼之间的离子化程度(pKa)差异,而非总体结构特征。数据还表明这两种药物共享一个共同受体,但与调节受体假说一致,它们通过由药物分子离子化程度决定的不同途径到达该受体。