Ragsdale D S, McPhee J C, Scheuer T, Catterall W A
Department of Pharmacology, University of Washington, Scattle 98195-7280, USA.
Proc Natl Acad Sci U S A. 1996 Aug 20;93(17):9270-5. doi: 10.1073/pnas.93.17.9270.
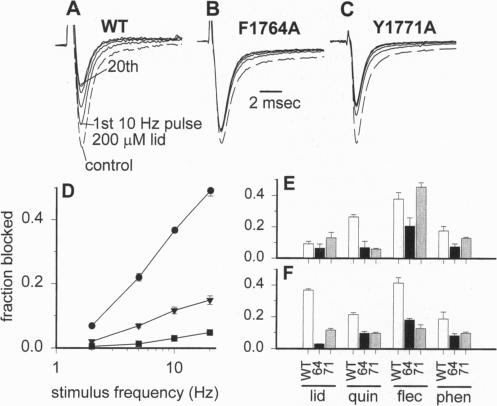
Voltage-gated Na+ channels are the molecular targets of local anesthetics, class I antiarrhythmic drugs, and some anticonvulsants. These chemically diverse drugs inhibit Na+ channels with complex voltage- and frequency-dependent properties that reflect preferential drug binding to open and inactivated channel states. The site-directed mutations F1764A and Y1771A in transmembrane segment IVS6 of type IIA Na+ channel alpha subunits dramatically reduce the affinity of inactivated channels for the local anesthetic etidocaine. In this study, we show that these mutations also greatly reduce the sensitivity of Na+ channels to state-dependent block by the class Ib antiarrhythmic drug lidocaine and the anticonvulsant phenytoin and, to a lesser extent, reduce the sensitivity to block by the class Ia and Ic antiarrhythmic drugs quinidine and flecainide. For lidocaine and phenytoin, which bind preferentially to inactivated Na+ channels, the mutation F1764A reduced the affinity for binding to the inactivated state 24.5-fold and 8.3-fold, respectively, while Y1771A had smaller effects. For quinidine and flecainide, which bind preferentially to the open Na+ channels, the mutations F1764A and Y1771A reduced the affinity for binding to the open state 2- to 3-fold. Thus, F1764 and Y1771 are common molecular determinants of state-dependent binding of diverse drugs including lidocaine, phenytoin, flecainide, and quinidine, suggesting that these drugs interact with a common receptor site. However, the different magnitude of the effects of these mutations on binding of the individual drugs indicates that they interact in an overlapping, but nonidentical, manner with a common receptor site. These results further define the contributions of F1764 and Y1771 to a complex drug receptor site in the pore of Na+ channels.
电压门控性钠通道是局部麻醉药、I类抗心律失常药物和一些抗惊厥药的分子靶点。这些化学结构多样的药物抑制钠通道,具有复杂的电压和频率依赖性特性,这反映了药物优先与开放和失活通道状态结合。IIA型钠通道α亚基跨膜片段IVS6中的定点突变F1764A和Y1771A显著降低了失活通道对局部麻醉药依替卡因的亲和力。在本研究中,我们表明这些突变还极大地降低了钠通道对Ib类抗心律失常药物利多卡因和抗惊厥药苯妥英钠的状态依赖性阻滞的敏感性,并且在较小程度上降低了对Ia类和Ic类抗心律失常药物奎尼丁和氟卡尼阻滞的敏感性。对于优先与失活钠通道结合的利多卡因和苯妥英钠,突变F1764A分别将与失活状态结合的亲和力降低了24.5倍和8.3倍,而Y1771A的影响较小。对于优先与开放钠通道结合的奎尼丁和氟卡尼,突变F1764A和Y1771A将与开放状态结合的亲和力降低了2至3倍。因此,F1764和Y1771是包括利多卡因、苯妥英钠、氟卡尼和奎尼丁在内的多种药物状态依赖性结合的共同分子决定因素,表明这些药物与一个共同的受体位点相互作用。然而,这些突变对个别药物结合影响的不同程度表明它们以重叠但不相同的方式与一个共同的受体位点相互作用。这些结果进一步明确了F1764和Y1771对钠通道孔中复杂药物受体位点的贡献。