Honys David, Twell David
Institute of Experimental Botany AS CR, Rozvojová 135, CZ-165 02, Praha 6, Czech Republic.
Genome Biol. 2004;5(11):R85. doi: 10.1186/gb-2004-5-11-r85. Epub 2004 Oct 27.
The haploid male gametophyte generation of flowering plants consists of two- or three-celled pollen grains. This functional specialization is thought to be a key factor in the evolutionary success of flowering plants. Moreover, pollen ontogeny is also an attractive model in which to dissect cellular networks that control cell growth, asymmetric cell division and cellular differentiation. Our objective, and an essential step towards the detailed understanding of these processes, was to comprehensively define the male haploid transcriptome throughout development.
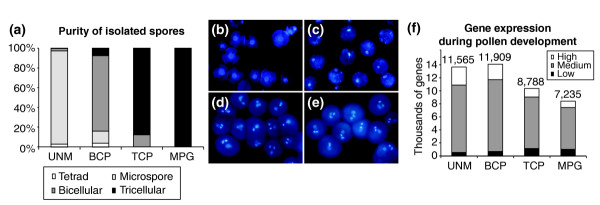
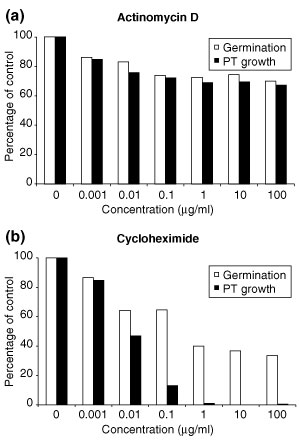
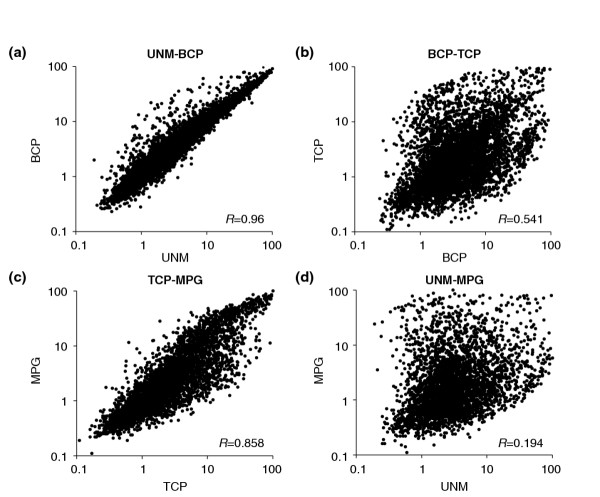
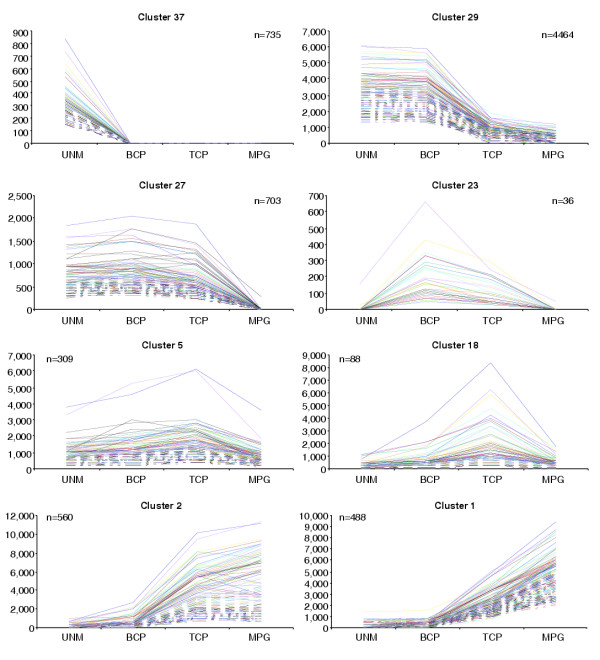
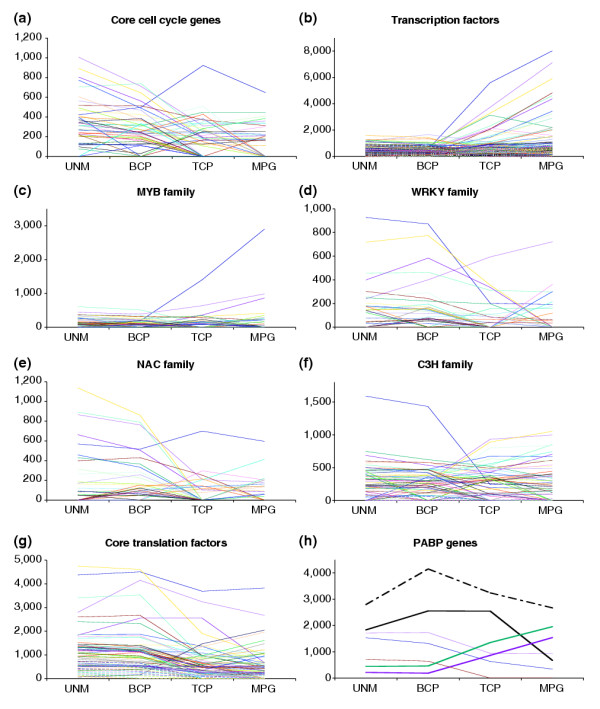
We have developed staged spore isolation procedures for Arabidopsis and used Affymetrix ATH1 genome arrays to identify a total of 13,977 male gametophyte-expressed mRNAs, 9.7% of which were male-gametophyte-specific. The transition from bicellular to tricellular pollen was accompanied by a decline in the number of diverse mRNA species and an increase in the proportion of male gametophyte-specific transcripts. Expression profiles of regulatory proteins and distinct clusters of coexpressed genes were identified that could correspond to components of gametophytic regulatory networks. Moreover, integration of transcriptome and experimental data revealed the early synthesis of translation factors and their requirement to support pollen tube growth.
The progression from proliferating microspores to terminally differentiated pollen is characterized by large-scale repression of early program genes and the activation of a unique late gene-expression program in maturing pollen. These data provide a quantum increase in knowledge concerning gametophytic transcription and lay the foundations for new genomic-led studies of the regulatory networks and cellular functions that operate to specify male gametophyte development.
开花植物的单倍体雄配子体世代由两细胞或三细胞花粉粒组成。这种功能特化被认为是开花植物进化成功的关键因素。此外,花粉个体发育也是剖析控制细胞生长、不对称细胞分裂和细胞分化的细胞网络的一个有吸引力的模型。我们的目标,也是详细了解这些过程的重要一步,是全面定义整个发育过程中的雄配子体转录组。
我们开发了拟南芥不同发育阶段孢子分离方法,并使用Affymetrix ATH1基因组芯片,共鉴定出13977个在雄配子体中表达的mRNA,其中9.7%是雄配子体特异的。从二细胞花粉到三细胞花粉的转变伴随着不同mRNA种类数量的减少以及雄配子体特异转录本比例的增加。我们鉴定出了调控蛋白的表达谱以及共表达基因的不同簇,它们可能对应于配子体调控网络的组成部分。此外,转录组和实验数据的整合揭示了翻译因子的早期合成及其对支持花粉管生长的需求。
从增殖的小孢子到终末分化花粉的过程,其特征是早期程序基因的大规模抑制以及成熟花粉中独特的晚期基因表达程序的激活。这些数据极大地增加了我们对配子体转录的了解,并为以新基因组学为导向的、对调控网络和细胞功能的研究奠定了基础,这些调控网络和细胞功能决定了雄配子体的发育。