MacLeod Dave, Charlebois Robert L, Doolittle Ford, Bapteste Eric
GenomeAtlantic, 1721 Lower Water Street, Suite 401, Halifax, NS, B3J 1S5, Canada.
BMC Evol Biol. 2005 Apr 8;5:27. doi: 10.1186/1471-2148-5-27.
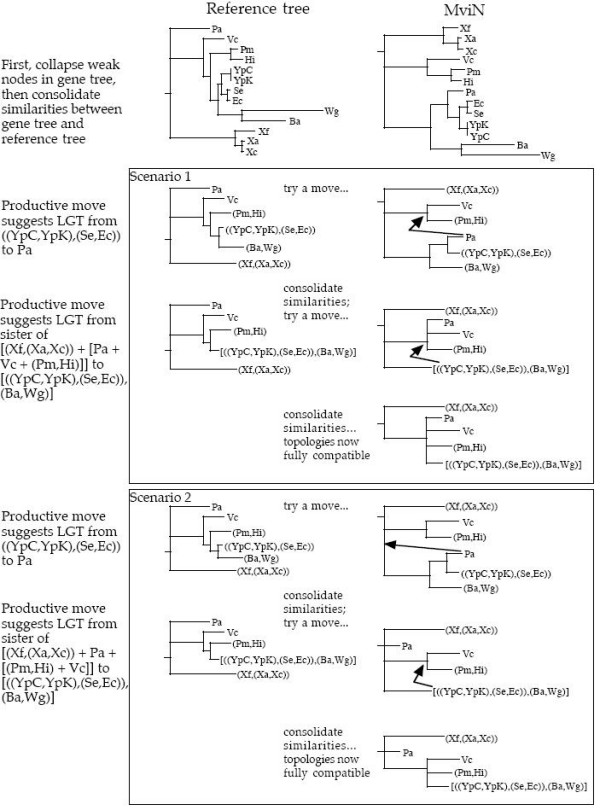
When organismal phylogenies based on sequences of single marker genes are poorly resolved, a logical approach is to add more markers, on the assumption that weak but congruent phylogenetic signal will be reinforced in such multigene trees. Such approaches are valid only when the several markers indeed have identical phylogenies, an issue which many multigene methods (such as the use of concatenated gene sequences or the assembly of supertrees) do not directly address. Indeed, even when the true history is a mixture of vertical descent for some genes and lateral gene transfer (LGT) for others, such methods produce unique topologies.
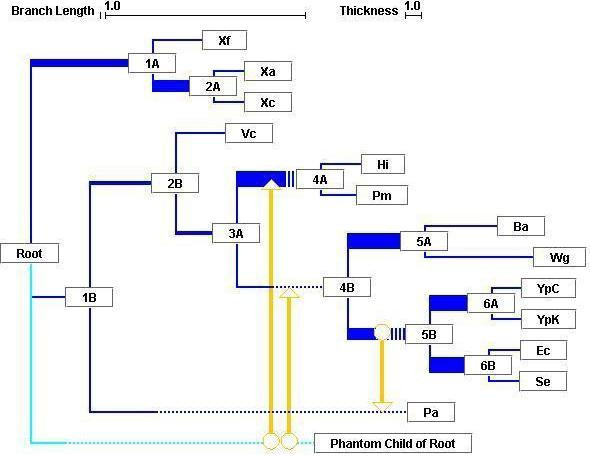
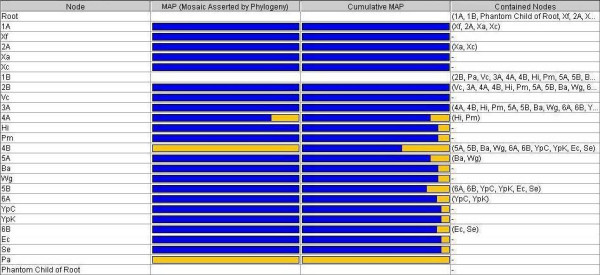
We have developed software that aims to extract evidence for vertical and lateral inheritance from a set of gene trees compared against an arbitrary reference tree. This evidence is then displayed as a synthesis showing support over the tree for vertical inheritance, overlaid with explicit lateral gene transfer (LGT) events inferred to have occurred over the history of the tree. Like splits-tree methods, one can thus identify nodes at which conflict occurs. Additionally one can make reasonable inferences about vertical and lateral signal, assigning putative donors and recipients.
A tool such as ours can serve to explore the reticulated dimensionality of molecular evolution, by dissecting vertical and lateral inheritance at high resolution. By this, we mean that individual nodes can be examined not only for congruence, but also for coherence in light of LGT. We assert that our tools will facilitate the comparison of phylogenetic trees, and the interpretation of conflicting data.
当基于单个标记基因序列构建的生物系统发育树分辨率较低时,一种合理的方法是添加更多标记,前提是在这种多基因树中微弱但一致的系统发育信号会得到加强。只有当几个标记确实具有相同的系统发育时,这种方法才有效,而许多多基因方法(如使用串联基因序列或组装超级树)并未直接解决这一问题。实际上,即使真实的历史是某些基因垂直遗传和其他基因横向基因转移(LGT)的混合,这些方法也会产生独特的拓扑结构。
我们开发了一款软件,旨在从与任意参考树进行比较的一组基因树中提取垂直和横向遗传的证据。然后,这些证据将以综合展示的形式呈现,显示树中垂直遗传的支持情况,并叠加推断在树的历史中发生的明确横向基因转移(LGT)事件。与分裂树方法一样,因此可以识别发生冲突的节点。此外,还可以对垂直和横向信号进行合理推断,确定假定的供体和受体。
像我们这样的工具可以通过高分辨率剖析垂直和横向遗传来探索分子进化的网状维度。我们的意思是,不仅可以检查各个节点的一致性,还可以根据LGT检查其连贯性。我们断言,我们的工具将有助于比较系统发育树,并解释冲突的数据。