Institute of Molecular Evolution, Heinrich-Heine-Universität Düsseldorf, Düsseldorf, Germany.
Genomic Microbiology Group, Institute of Microbiology, Christian-Albrechts-Universität Kiel, Kiel, Germany.
BMC Evol Biol. 2014 Dec 30;14:266. doi: 10.1186/s12862-014-0266-0.
Analyzed individually, gene trees for a given taxon set tend to harbour incongruent or conflicting signals. One popular approach to deal with this circumstance is to use concatenated data. But especially in prokaryotes, where lateral gene transfer (LGT) is a natural mechanism of generating genetic diversity, there are open questions as to whether concatenation amplifies or averages phylogenetic signals residing in individual genes. Here we investigate concatenations of prokaryotic and eukaryotic datasets to investigate possible sources of incongruence in phylogenetic trees and to examine the level of overlap between individual and concatenated alignments.
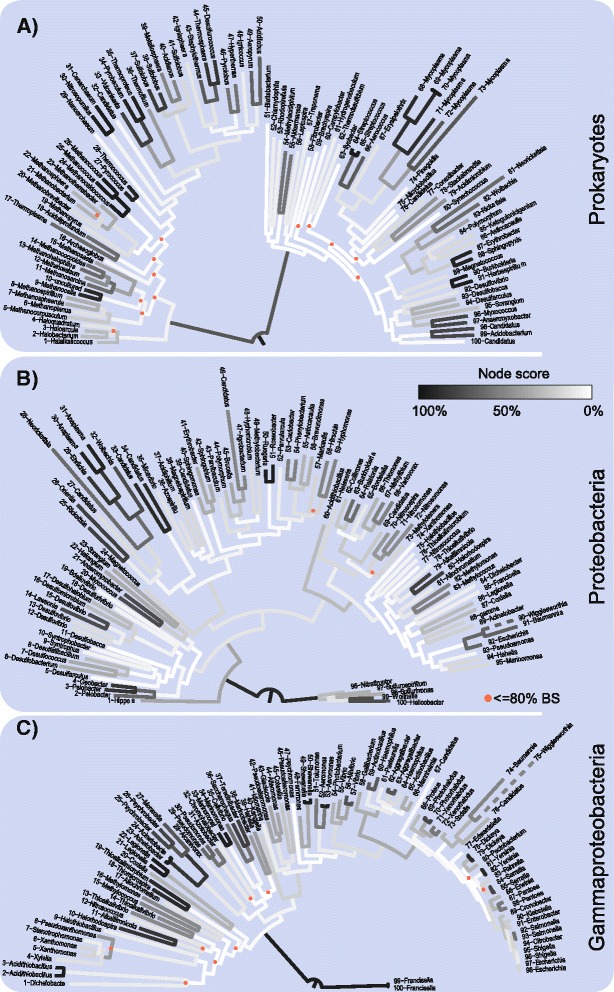
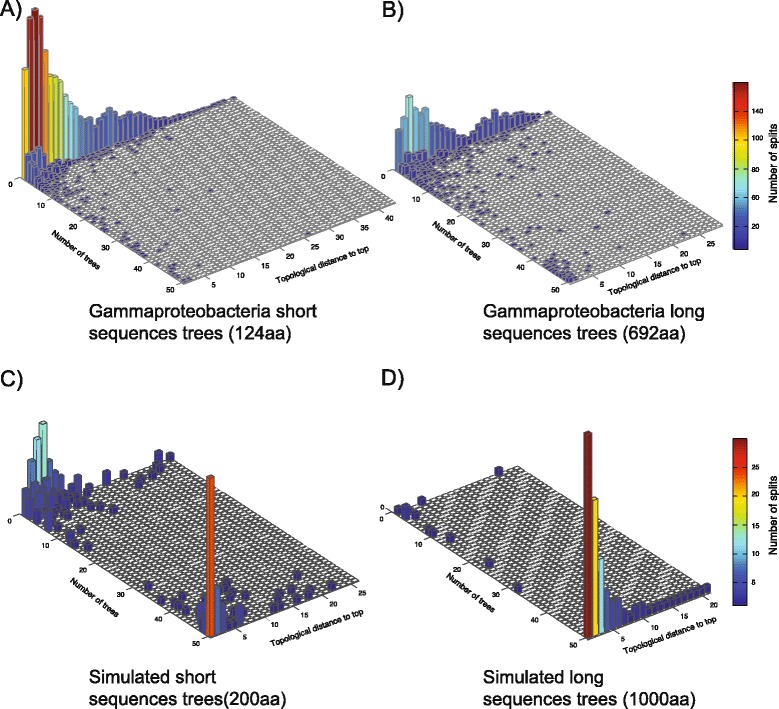
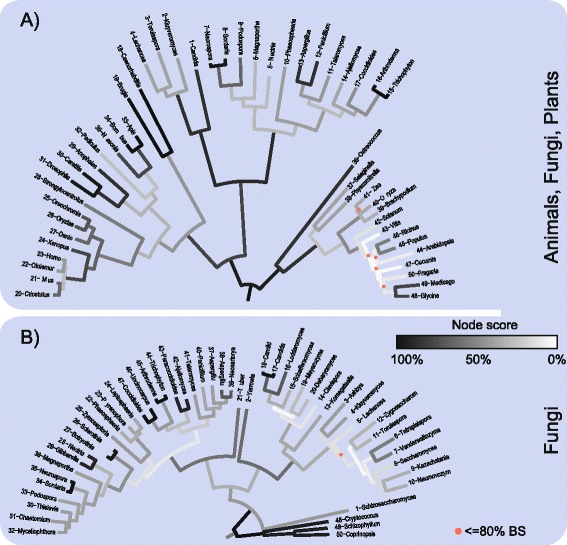
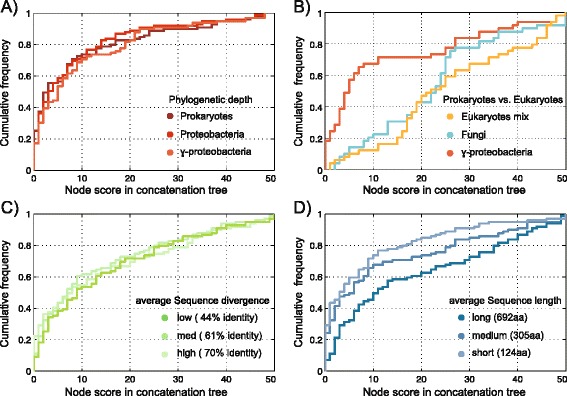
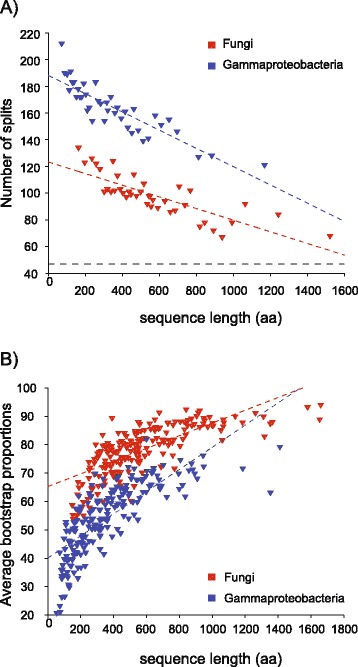
We analyzed prokaryotic datasets comprising 248 invidual gene trees from 315 genomes at three taxonomic depths spanning gammaproteobacteria, proteobacteria, and prokaryotes (bacteria plus archaea), and eukaryotic datasets comprising 279 invidual gene trees from 85 genomes at two taxonomic depths: across plants-animals-fungi and within fungi. Consistent with previous findings, the branches in trees made from concatenated alignments are, in general, not supported by any of their underlying individual gene trees, even though the concatenation trees tend to possess high bootstrap proportions values. For the prokaryote data, this observation is independent of phylogenetic depth and sequence conservation. The eukaryotic data show much better agreement between concatenation and single gene trees. LGT frequencies in trees were estimated using established methods. Sequence length in individual alignments, but not sequence divergence, was found to correlate with the generation of branches that correspond to the concatenated tree.
The weak correspondence of concatenation trees with single gene trees gives rise to the question where the phylogenetic signal in concatenated trees is coming from. The eukaryote data reveals a better correspondence between individual and concatenation trees than the prokaryote data. The question of whether the lack of correspondence between individual genes and the concatenation tree in the prokaryotic data is due to LGT or phylogenetic artefacts remains unanswered. If LGT is the cause of incongruence between concatenation and individual trees, we would have expected to see greater degrees of incongruence for more divergent prokaryotic data sets, which was not observed, although estimated rates of LGT suggest that LGT is responsible for at least some of the observed incongruence.
分析给定分类单元集的基因树时,往往会存在不一致或冲突的信号。一种常用的处理方法是使用串联数据。但是,特别是在原核生物中,侧向基因转移(LGT)是产生遗传多样性的自然机制,因此对于串联是否会放大或平均存在于单个基因中的系统发育信号存在一些问题。在这里,我们研究了原核生物和真核生物数据集的串联,以调查系统发育树中不一致的可能来源,并检查单个和串联比对之间的重叠程度。
我们分析了由 315 个基因组中的 248 个个体基因树组成的原核生物数据集,涵盖了γ变形菌门、变形菌门和原核生物(细菌加古菌)三个分类深度,以及由 85 个基因组中的 279 个个体基因树组成的真核生物数据集,涵盖了植物-动物-真菌和真菌内部两个分类深度。与先前的发现一致,即使串联树倾向于具有较高的自举比例值,但来自串联比对的树的分支通常不受其基础的任何单个基因树的支持。对于原核生物数据,这种观察结果与系统发育深度和序列保守性无关。真核生物数据显示串联和单个基因树之间的一致性要好得多。使用已建立的方法估计了树中的 LGT 频率。在单个比对中的序列长度,而不是序列差异,与对应于串联树的分支的产生相关。
串联树与单个基因树之间的弱对应性引发了一个问题,即串联树中的系统发育信号来自何处。与原核生物数据相比,真核生物数据显示出单个和串联树之间更好的对应关系。原核生物数据中,单个基因与串联树之间的对应关系缺失是否由于 LGT 或系统发育伪影引起的问题仍未得到解答。如果 LGT 是串联和单个树之间不一致的原因,我们预计更具分歧的原核生物数据集会出现更大程度的不一致性,但这种情况并未观察到,尽管估计的 LGT 率表明 LGT 至少是导致一些观察到的不一致性的原因之一。