McLaughlin Stuart, Smith Steven O, Hayman Michael J, Murray Diana
Department of Physiology and Biophysics, HSC, Stony Brook University, Stony Brook, NY 11794, USA.
J Gen Physiol. 2005 Jul;126(1):41-53. doi: 10.1085/jgp.200509274. Epub 2005 Jun 13.
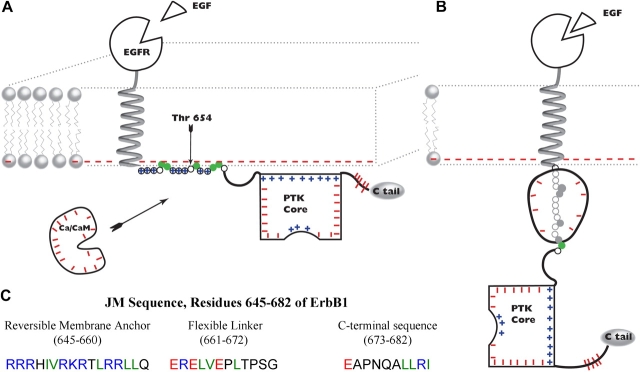
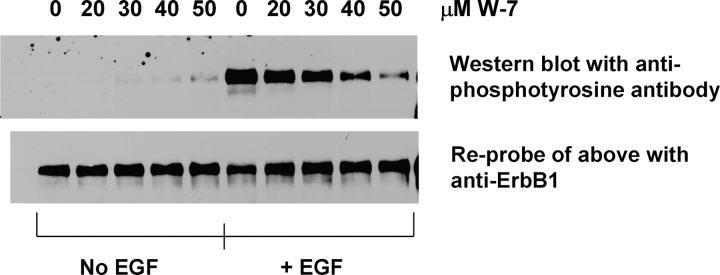
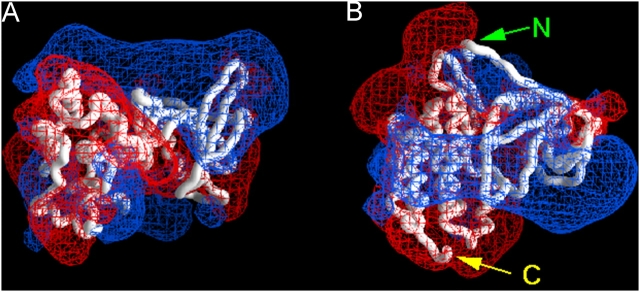
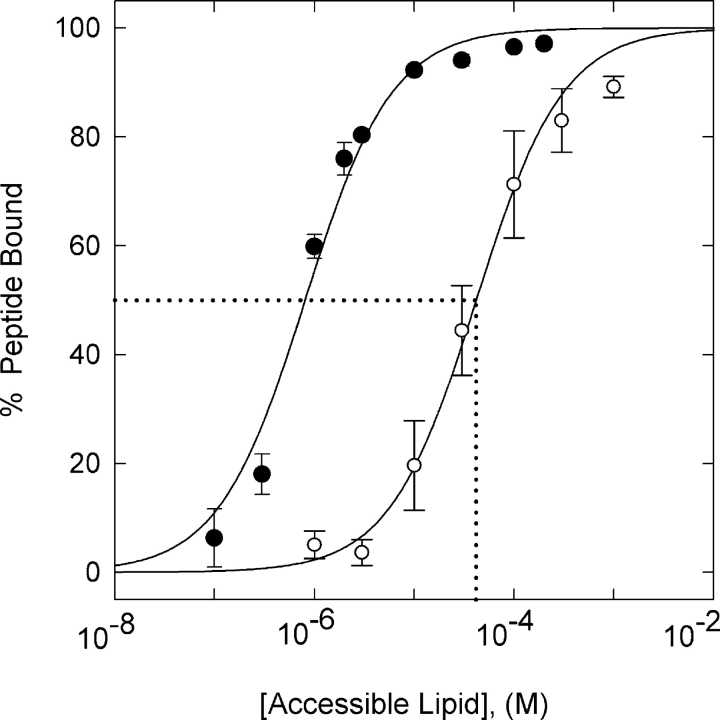
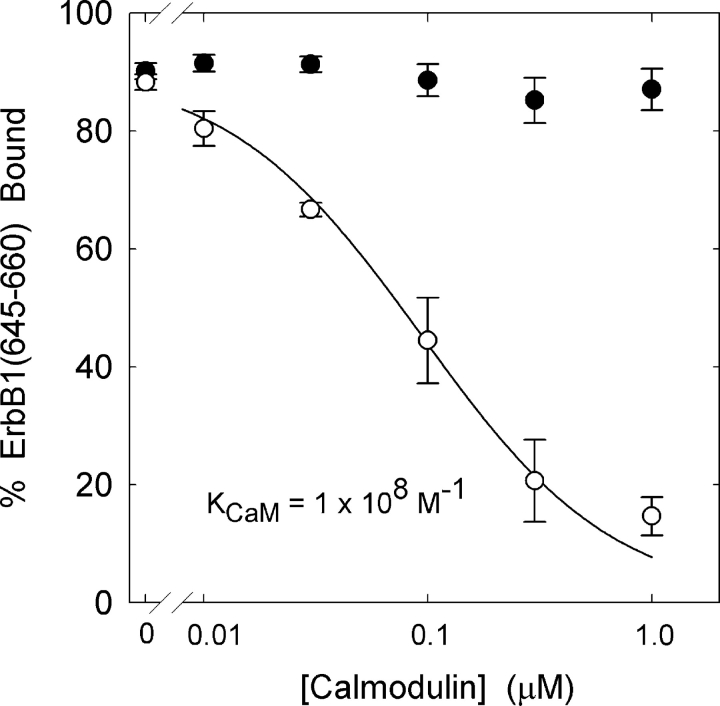
We propose a new mechanism to explain autoinhibition of the epidermal growth factor receptor (EGFR/ErbB) family of receptor tyrosine kinases based on a structural model that postulates both their juxtamembrane and protein tyrosine kinase domains bind electrostatically to acidic lipids in the plasma membrane, restricting access of the kinase domain to substrate tyrosines. Ligand-induced dimerization promotes partial trans autophosphorylation of ErbB1, leading to a rapid rise in intracellular [Ca(2+)] that can activate calmodulin. We postulate the Ca(2+)/calmodulin complex binds rapidly to residues 645--660 of the juxtamembrane domain, reversing its net charge from +8 to -8 and repelling it from the negatively charged inner leaflet of the membrane. The repulsion has two consequences: it releases electrostatically sequestered phosphatidylinositol 4,5-bisphosphate (PIP(2)), and it disengages the kinase domain from the membrane, allowing it to become fully active and phosphorylate an adjacent ErbB molecule or other substrate. We tested various aspects of the model by measuring ErbB juxtamembrane peptide binding to phospholipid vesicles using both a centrifugation assay and fluorescence correlation spectroscopy; analyzing the kinetics of interactions between ErbB peptides, membranes, and Ca(2+)/calmodulin using fluorescence stop flow; assessing ErbB1 activation in Cos1 cells; measuring fluorescence resonance energy transfer between ErbB peptides and PIP(2); and making theoretical electrostatic calculations on atomic models of membranes and ErbB juxtamembrane and kinase domains.
我们提出了一种新机制来解释受体酪氨酸激酶表皮生长因子受体(EGFR/ErbB)家族的自抑制作用,该机制基于一个结构模型,该模型假定其近膜结构域和蛋白酪氨酸激酶结构域均通过静电作用与质膜中的酸性脂质结合,从而限制激酶结构域与底物酪氨酸的接触。配体诱导的二聚化促进了ErbB1的部分反式自磷酸化,导致细胞内[Ca(2+)]迅速升高,进而激活钙调蛋白。我们推测Ca(2+)/钙调蛋白复合物会迅速与近膜结构域的645-660位残基结合,使其净电荷从+8变为-8,并将其从带负电荷的膜内小叶排斥开。这种排斥有两个后果:它释放了通过静电作用被隔离的磷脂酰肌醇4,5-二磷酸(PIP(2)),并且使激酶结构域与膜分离,使其能够完全激活并磷酸化相邻的ErbB分子或其他底物。我们通过以下方式测试了该模型的各个方面:使用离心测定法和荧光相关光谱法测量ErbB近膜肽与磷脂囊泡的结合;使用荧光停流分析法分析ErbB肽、膜和Ca(2+)/钙调蛋白之间相互作用的动力学;评估Cos1细胞中ErbB1的激活情况;测量ErbB肽与PIP(2)之间的荧光共振能量转移;以及对膜、ErbB近膜结构域和激酶结构域的原子模型进行理论静电计算。