Banks Jay L, Beard Hege S, Cao Yixiang, Cho Art E, Damm Wolfgang, Farid Ramy, Felts Anthony K, Halgren Thomas A, Mainz Daniel T, Maple Jon R, Murphy Robert, Philipp Dean M, Repasky Matthew P, Zhang Linda Y, Berne Bruce J, Friesner Richard A, Gallicchio Emilio, Levy Ronald M
Schrödinger, Inc., New York, NY 10036, USA.
J Comput Chem. 2005 Dec;26(16):1752-80. doi: 10.1002/jcc.20292.
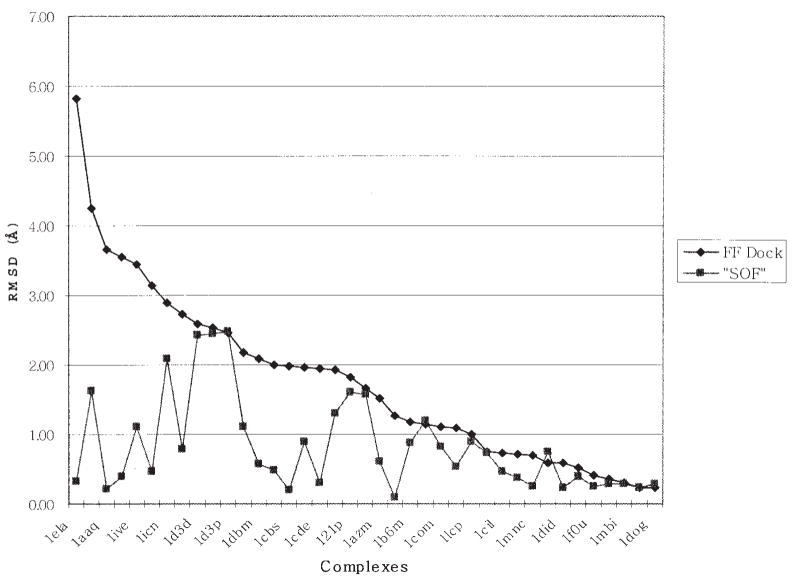
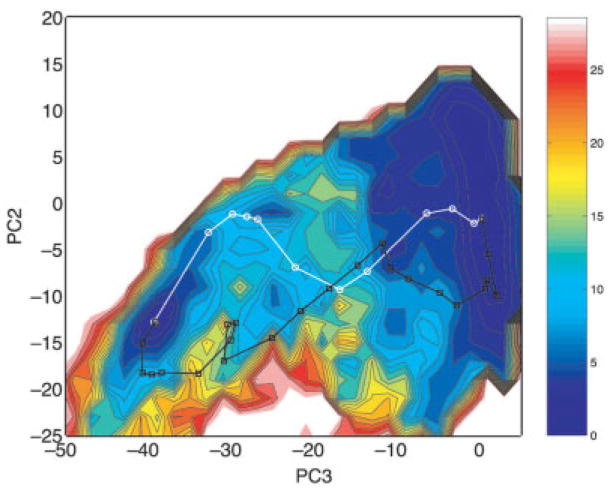
We provide an overview of the IMPACT molecular mechanics program with an emphasis on recent developments and a description of its current functionality. With respect to core molecular mechanics technologies we include a status report for the fixed charge and polarizable force fields that can be used with the program and illustrate how the force fields, when used together with new atom typing and parameter assignment modules, have greatly expanded the coverage of organic compounds and medicinally relevant ligands. As we discuss in this review, explicit solvent simulations have been used to guide our design of implicit solvent models based on the generalized Born framework and a novel nonpolar estimator that have recently been incorporated into the program. With IMPACT it is possible to use several different advanced conformational sampling algorithms based on combining features of molecular dynamics and Monte Carlo simulations. The program includes two specialized molecular mechanics modules: Glide, a high-throughput docking program, and QSite, a mixed quantum mechanics/molecular mechanics module. These modules employ the IMPACT infrastructure as a starting point for the construction of the protein model and assignment of molecular mechanics parameters, but have then been developed to meet specialized objectives with respect to sampling and the energy function.
我们概述了IMPACT分子力学程序,重点介绍了其近期的发展情况,并描述了其当前功能。关于核心分子力学技术,我们提供了可与该程序一起使用的固定电荷和可极化力场的现状报告,并说明了当力场与新的原子类型和参数分配模块一起使用时,如何极大地扩展了有机化合物和与药物相关配体的覆盖范围。正如我们在本综述中所讨论的,显式溶剂模拟已被用于指导我们基于广义玻恩框架和一种新型非极性估计器设计隐式溶剂模型,这些模型最近已被纳入该程序。使用IMPACT,可以基于分子动力学和蒙特卡罗模拟的组合特征使用几种不同的先进构象采样算法。该程序包括两个专门的分子力学模块:Glide,一个高通量对接程序;以及QSite,一个混合量子力学/分子力学模块。这些模块以IMPACT基础设施为起点构建蛋白质模型并分配分子力学参数,但随后经过开发以满足关于采样和能量函数的特定目标。