Indap Amit R, Marth Gabor T, Struble Craig A, Tonellato Peter, Olivier Michael
Human and Molecular Genetics Center, Medical College of Wisconsin, Milwaukee, USA.
BMC Bioinformatics. 2005 Dec 15;6:303. doi: 10.1186/1471-2105-6-303.
Different classes of haplotype block algorithms exist and the ideal dataset to assess their performance would be to comprehensively re-sequence a large genomic region in a large population. Such data sets are expensive to collect. Alternatively, we performed coalescent simulations to generate haplotypes with a high marker density and compared block partitioning results from diversity based, LD based, and information theoretic algorithms under different values of SNP density and allele frequency.
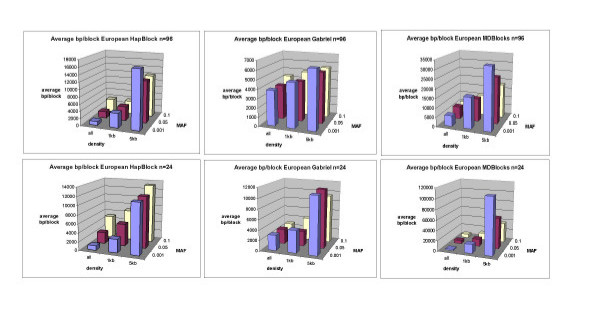
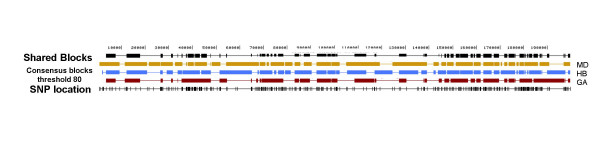
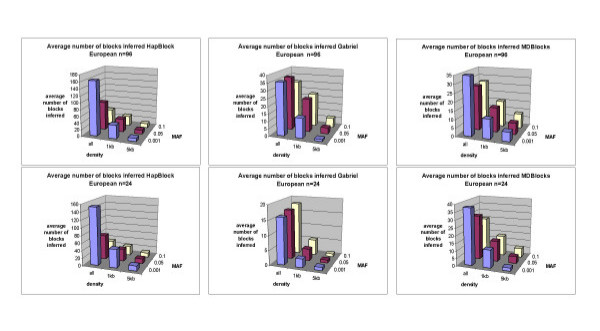
We simulated 1000 haplotypes using the standard coalescent for three world populations--European, African American, and East Asian--and applied three classes of block partitioning algorithms--diversity based, LD based, and information theoretic. We assessed algorithm differences in number, size, and coverage of blocks inferred under different conditions of SNP density, allele frequency, and sample size. Each algorithm inferred blocks differing in number, size, and coverage under different density and allele frequency conditions. Different partitions had few if any matching block boundaries. However they still overlapped and a high percentage of total chromosomal region was common to all methods. This percentage was generally higher with a higher density of SNPs and when rarer markers were included.
A gold standard definition of a haplotype block is difficult to achieve, but collecting haplotypes covered with a high density of SNPs, partitioning them with a variety of block algorithms, and identifying regions common to all methods may be the best way to identify genomic regions that harbor SNP variants that cause disease.
存在不同类别的单倍型块算法,而评估其性能的理想数据集是在大量人群中对大的基因组区域进行全面重测序。收集这样的数据集成本很高。作为替代方案,我们进行了合并模拟以生成具有高标记密度的单倍型,并比较了基于多样性、基于连锁不平衡(LD)和信息论算法在不同单核苷酸多态性(SNP)密度和等位基因频率值下的块划分结果。
我们使用标准合并方法为欧洲、非裔美国人和东亚这三个人群模拟了1000个单倍型,并应用了三类块划分算法——基于多样性、基于LD和信息论算法。我们评估了在SNP密度、等位基因频率和样本量的不同条件下推断出的块在数量、大小和覆盖范围方面的算法差异。在不同的密度和等位基因频率条件下,每种算法推断出的块在数量、大小和覆盖范围上都有所不同。不同的划分几乎没有匹配的块边界。然而它们仍然有重叠,并且所有方法共有的总染色体区域的百分比很高。当SNP密度较高且包含较罕见的标记时,这个百分比通常更高。
单倍型块的金标准定义很难实现,但收集覆盖高密度SNP的单倍型,用多种块算法对其进行划分,并识别所有方法共有的区域,可能是识别含有导致疾病的SNP变异的基因组区域的最佳方法。