Levitt David G, Schoemaker Rik C
Department of Physiology, University of Minnesota, Minneapolis, MN 55455, USA.
BMC Clin Pharmacol. 2006 Jan 6;6:1. doi: 10.1186/1472-6904-6-1.
The angiotensin-converting enzyme (ACE) inhibitors have complicated and poorly characterized pharmacokinetics. There are two binding sites per ACE (high affinity "C", lower affinity "N") that have sub-nanomolar affinities and dissociation rates of hours. Most inhibitors are given orally in a prodrug form that is systemically converted to the active form. This paper describes the first human physiologically based pharmacokinetic (PBPK) model of this drug class.
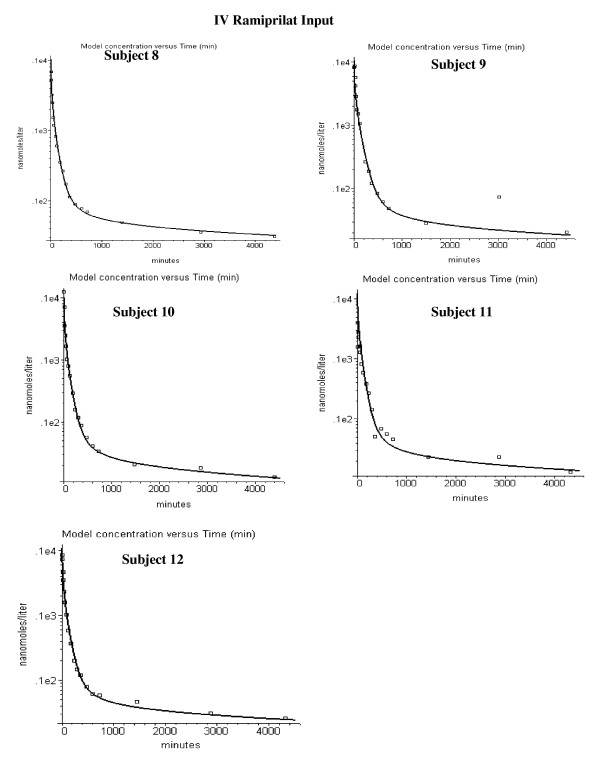
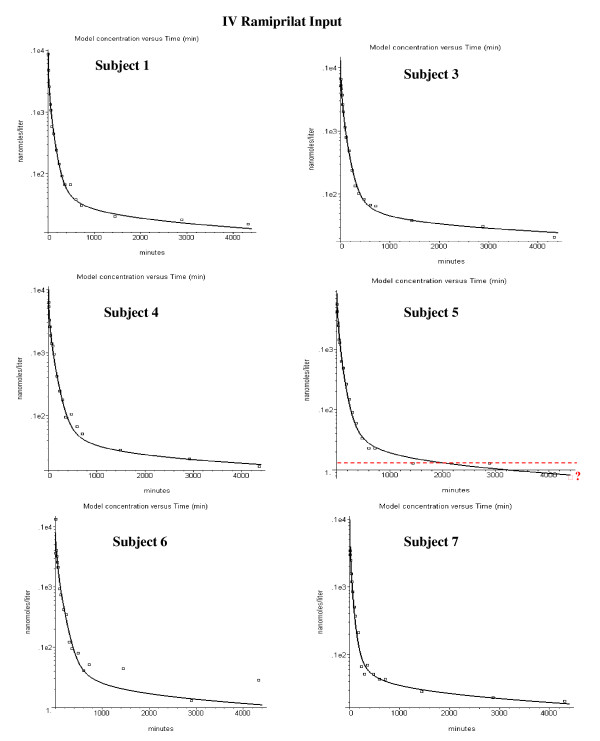
The model was applied to the experimental data of van Griensven et. al for the pharmacokinetics of ramiprilat and its prodrug ramipril. It describes the time course of the inhibition of the N and C ACE sites in plasma and the different tissues. The model includes: 1) two independent ACE binding sites; 2) non-equilibrium time dependent binding; 3) liver and kidney ramipril intracellular uptake, conversion to ramiprilat and extrusion from the cell; 4) intestinal ramipril absorption. The experimental in vitro ramiprilat/ACE binding kinetics at 4 degrees C and 300 mM NaCl were assumed for most of the PBPK calculations. The model was incorporated into the freely distributed PBPK program PKQuest.
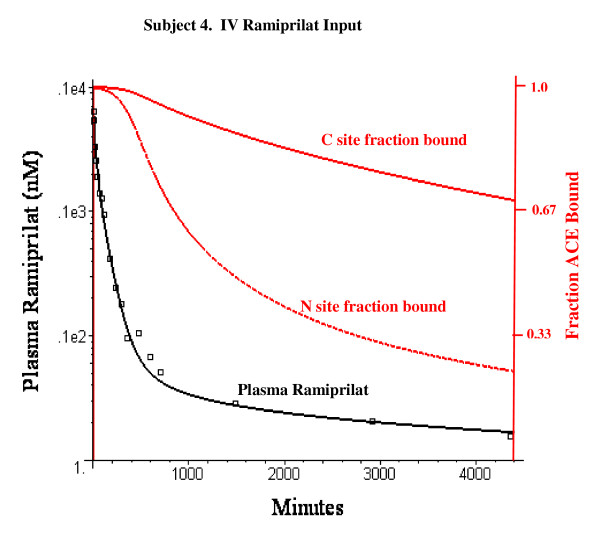
The PBPK model provides an accurate description of the individual variation of the plasma ramipril and ramiprilat and the ramiprilat renal clearance following IV ramiprilat and IV and oral ramipril. Summary of model features: Less than 2% of total body ACE is in plasma; 35% of the oral dose is absorbed; 75% of the ramipril metabolism is hepatic and 25% of this is converted to systemic ramiprilat; 100% of renal ramipril metabolism is converted to systemic ramiprilat. The inhibition was long lasting, with 80% of the C site and 33% of the N site inhibited 24 hours following a 2.5 mg oral ramipril dose. The plasma ACE inhibition determined by the standard assay is significantly less than the true in vivo inhibition because of assay dilution.
If the in vitro plasma binding kinetics of the ACE inhibitor for the two binding sites are known, a unique PBPK model description of the Griensven et. al. experimental data can be obtained.
血管紧张素转换酶(ACE)抑制剂具有复杂且特征不明的药代动力学。每个ACE有两个结合位点(高亲和力的“C”位点和低亲和力的“N”位点),其亲和力低于纳摩尔级别,解离速率为数小时。大多数抑制剂以前药形式口服给药,前药在体内会系统地转化为活性形式。本文描述了该类药物首个基于人体生理学的药代动力学(PBPK)模型。
该模型应用于van Griensven等人关于雷米普利拉及其前药雷米普利药代动力学的实验数据。它描述了血浆和不同组织中N和C ACE位点抑制作用的时间进程。该模型包括:1)两个独立的ACE结合位点;2)非平衡时间依赖性结合;3)肝脏和肾脏中雷米普利的细胞内摄取、转化为雷米普利拉并从细胞中排出;4)肠道对雷米普利的吸收。在大多数PBPK计算中,假定在4℃和300 mM NaCl条件下的雷米普利拉/ACE体外结合动力学实验数据。该模型被纳入免费分发的PBPK程序PKQuest中。
PBPK模型准确描述了静脉注射雷米普利拉以及静脉注射和口服雷米普利后血浆中雷米普利和雷米普利拉的个体差异以及雷米普利拉的肾脏清除率。模型特征总结:全身ACE总量中不到2%存在于血浆中;口服剂量的35%被吸收;雷米普利代谢的75%发生在肝脏,其中25%转化为全身的雷米普利拉;肾脏中雷米普利代谢的100%转化为全身的雷米普利拉。抑制作用持续时间长,口服2.5 mg雷米普利后24小时,80%的C位点和33%的N位点被抑制。由于检测稀释,标准检测方法测定的血浆ACE抑制作用明显低于体内真实的抑制作用。
如果已知ACE抑制剂在两个结合位点的体外血浆结合动力学,就可以获得对van Griensven等人实验数据的独特PBPK模型描述。