Zhang Zhang, Li Jun, Yu Jun
Institute of Computing Technology, Chinese Academy of Sciences, Beijing 100080, China.
BMC Evol Biol. 2006 Jun 2;6:44. doi: 10.1186/1471-2148-6-44.
Approximate methods for estimating nonsynonymous and synonymous substitution rates (Ka and Ks) among protein-coding sequences have adopted different mutation (substitution) models. In the past two decades, several methods have been proposed but they have not considered unequal transitional substitutions (between the two purines, A and G, or the two pyrimidines, T and C) that become apparent when sequences data to be compared are vast and significantly diverged.
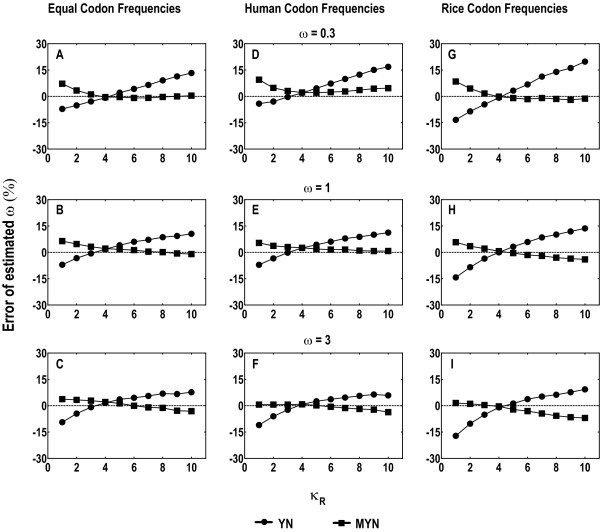
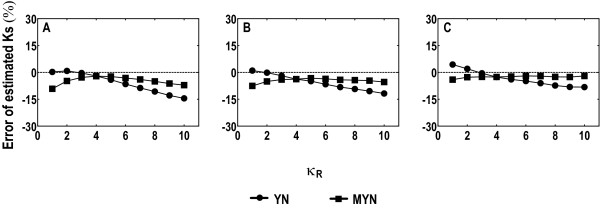
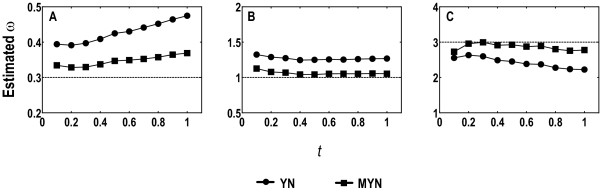
We propose a new method (MYN), a modified version of the Yang-Nielsen algorithm (YN), for evolutionary analysis of protein-coding sequences in general. MYN adopts the Tamura-Nei Model that considers the difference among rates of transitional and transversional substitutions as well as factors in codon frequency bias. We evaluate the performance of MYN by comparing to other methods, especially to YN, and to show that MYN has minimal deviations when parameters vary within normal ranges defined by empirical data.
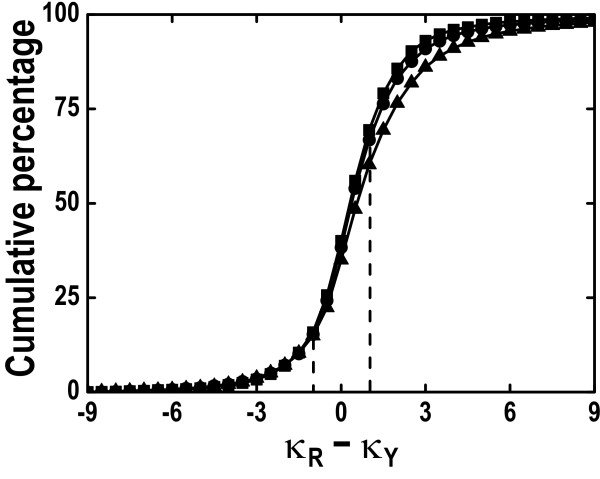
Our comparative results deriving from consistency analysis, computer simulations and authentic datasets, indicate that ignoring unequal transitional rates may lead to serious biases and that MYN performs well in most of the tested cases. These results also suggest that acquisitions of reliable synonymous and nonsynonymous substitution rates primarily depend on less biased estimates of transition/transversion rate ratio.
用于估计蛋白质编码序列中非同义替换率和同义替换率(Ka和Ks)的近似方法采用了不同的突变(替换)模型。在过去二十年中,已经提出了几种方法,但当要比较的序列数据量巨大且差异显著时,它们没有考虑到明显的不等同转换替换(在两个嘌呤A和G之间,或两个嘧啶T和C之间)。
我们提出了一种新方法(MYN),它是杨 - 尼尔森算法(YN)的改进版本,用于一般蛋白质编码序列的进化分析。MYN采用了塔穆拉 - 内模型,该模型考虑了转换和颠换替换率之间的差异以及密码子频率偏差等因素。我们通过与其他方法(特别是YN)进行比较来评估MYN的性能,并表明当参数在由经验数据定义的正常范围内变化时,MYN的偏差最小。
我们从一致性分析、计算机模拟和真实数据集得出的比较结果表明,忽略不等同转换率可能会导致严重偏差,并且MYN在大多数测试案例中表现良好。这些结果还表明,获得可靠的同义替换率和非同义替换率主要取决于对转换/颠换率比的偏差较小的估计。