Carro Angel, Tress Michael, de Juan David, Pazos Florencio, Lopez-Romero Pedro, del Sol Antonio, Valencia Alfonso, Rojas Ana M
Protein Design Group, CNB-CSIC C/Darwin n.3, 28049, Madrid, Spain.
Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W110-5. doi: 10.1093/nar/gkl203.
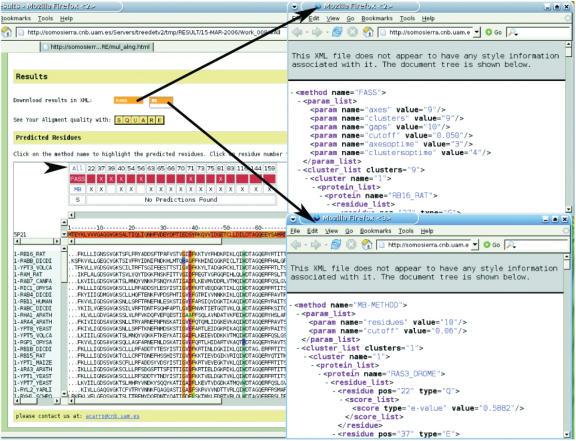
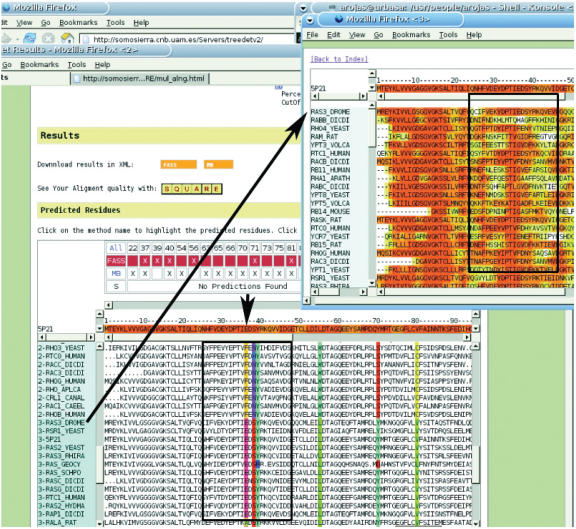
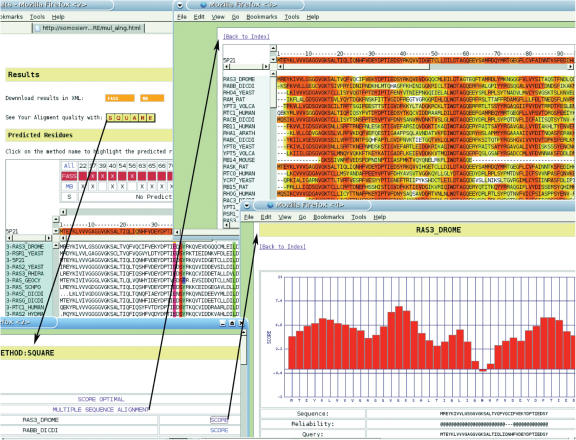
The TreeDet (Tree Determinant) Server is the first release of a system designed to integrate results from methods that predict functional sites in protein families. These methods take into account the relation between sequence conservation and evolutionary importance. TreeDet fully analyses the space of protein sequences in either user-uploaded or automatically generated multiple sequence alignments. The methods implemented in the server represent three main classes of methods for the detection of family-dependent conserved positions, a tree-based method, a correlation based method and a method that employs a principal component analyses coupled to a cluster algorithm. An additional method is provided to highlight the reliability of the position in the alignments. The server is available at http://www.pdg.cnb.uam.es/servers/treedet.
TreeDet(树状决定因素)服务器是一个系统的首个版本,该系统旨在整合来自预测蛋白质家族功能位点方法的结果。这些方法考虑了序列保守性与进化重要性之间的关系。TreeDet会全面分析用户上传的或自动生成的多序列比对中的蛋白质序列空间。服务器中实现的方法代表了用于检测家族依赖性保守位置的三类主要方法,一种基于树的方法、一种基于相关性的方法以及一种将主成分分析与聚类算法相结合的方法。还提供了一种额外的方法来突出比对中位置的可靠性。该服务器可在http://www.pdg.cnb.uam.es/servers/treedet获取。