Biro Jan Charles
Homulus Foundation, San Francisco, CA 94105, USA.
Theor Biol Med Model. 2006 Aug 7;3:28. doi: 10.1186/1742-4682-3-28.
All the information necessary for protein folding is supposed to be present in the amino acid sequence. It is still not possible to provide specific ab initio structure predictions by bioinformatical methods. It is suspected that additional folding information is present in protein coding nucleic acid sequences, but this is not represented by the known genetic code.
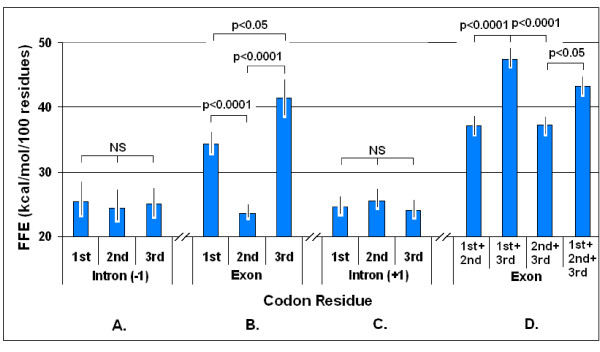
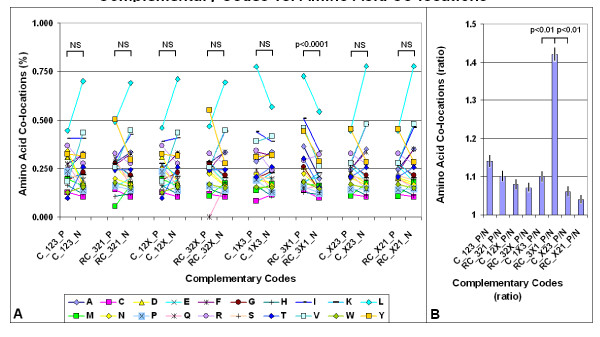
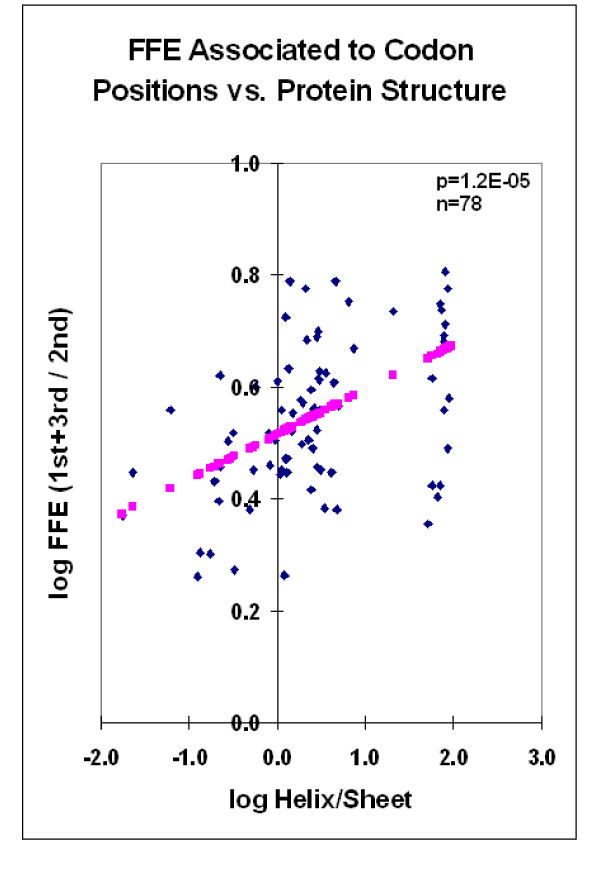
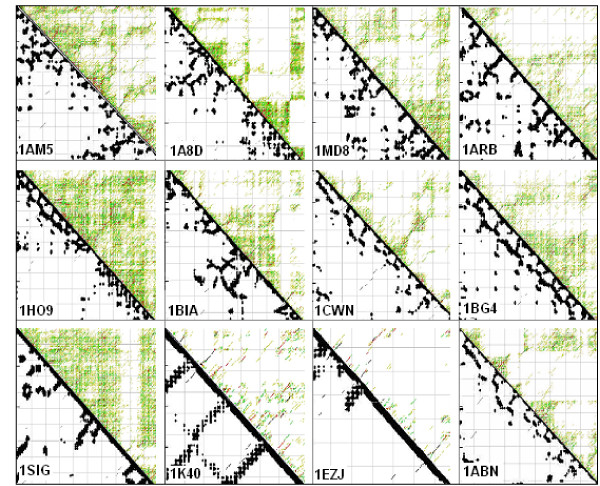
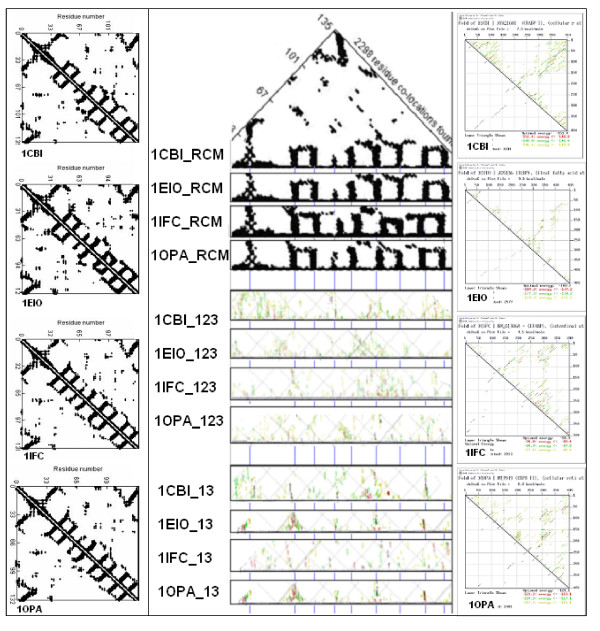
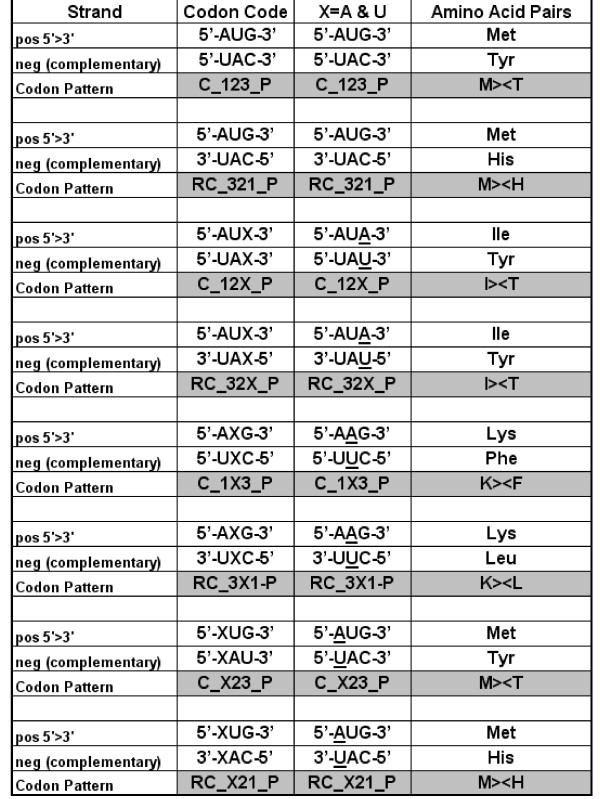
Nucleic acid subsequences comprising the 1st and/or 3rd codon residues in mRNAs express significantly higher free folding energy (FFE) than the subsequence containing only the 2nd residues (p < 0.0001, n = 81). This periodic FFE difference is not present in introns. It is therefore a specific physico-chemical characteristic of coding sequences and might contribute to unambiguous definition of codon boundaries during translation. The FFEs of the 1st and 3rd residues are additive, which suggests that these residues contain a significant number of complementary bases and that may contribute to selection for local RNA secondary structures in coding regions. This periodic, codon-related structure-formation of mRNAs indicates a connection between the structures of exons and the corresponding (translated) proteins. The folding energy dot plots of RNAs and the residue contact maps of the coded proteins are indeed similar. Residue contact statistics using 81 different protein structures confirmed that amino acids that are coded by partially reverse and complementary codons (Watson-Crick (WC) base pairs at the 1st and 3rd codon positions and translated in reverse orientation) are preferentially co-located in protein structures.
Exons are distinguished from introns, and codon boundaries are physico-chemically defined, by periodically distributed FFE differences between codon positions. There is a selection for local RNA secondary structures in coding regions and this nucleic acid structure resembles the folding profiles of the coded proteins. The preferentially (specifically) interacting amino acids are coded by partially complementary codons, which strongly supports the connection between mRNA and the corresponding protein structures and indicates that there is protein folding information in nucleic acids that is not present in the genetic code. This might suggest an additional explanation of codon redundancy.
蛋白质折叠所需的所有信息都应该存在于氨基酸序列中。目前仍无法通过生物信息学方法进行特定的从头结构预测。有人怀疑蛋白质编码核酸序列中存在额外的折叠信息,但这并未由已知的遗传密码所体现。
包含mRNA中第1和/或第3密码子残基的核酸子序列表达出的自由折叠能(FFE)明显高于仅包含第2残基的子序列(p < 0.0001,n = 81)。这种周期性的FFE差异在内含子中不存在。因此,这是编码序列的一种特定物理化学特征,可能有助于在翻译过程中明确界定密码子边界。第1和第3残基的FFE是可加的,这表明这些残基包含大量互补碱基,可能有助于选择编码区域中的局部RNA二级结构。这种mRNA的周期性、与密码子相关的结构形成表明外显子结构与相应(翻译后的)蛋白质之间存在联系。RNA的折叠能点阵图和编码蛋白质的残基接触图确实相似。使用81种不同蛋白质结构的残基接触统计证实,由部分反向互补密码子编码的氨基酸(第1和第3密码子位置为沃森-克里克(WC)碱基对且反向翻译)在蛋白质结构中优先共定位。
外显子与内含子不同,密码子边界通过密码子位置之间周期性分布的FFE差异在物理化学上得以界定。编码区域中存在对局部RNA二级结构的选择,且这种核酸结构类似于编码蛋白质的折叠轮廓。优先(特异性)相互作用的氨基酸由部分互补密码子编码,这有力地支持了mRNA与相应蛋白质结构之间的联系,并表明核酸中存在遗传密码中不存在的蛋白质折叠信息。这可能为密码子冗余提供额外的解释。