Fredslund Jakob, Madsen Lene H, Hougaard Birgit K, Nielsen Anna Marie, Bertioli David, Sandal Niels, Stougaard Jens, Schauser Leif
Bioinformatics Research Center, University of Aarhus, Høegh-Guldbergs Gade 10, Building 090, DK-8000 Arhus C, Denmark.
BMC Genomics. 2006 Aug 14;7:207. doi: 10.1186/1471-2164-7-207.
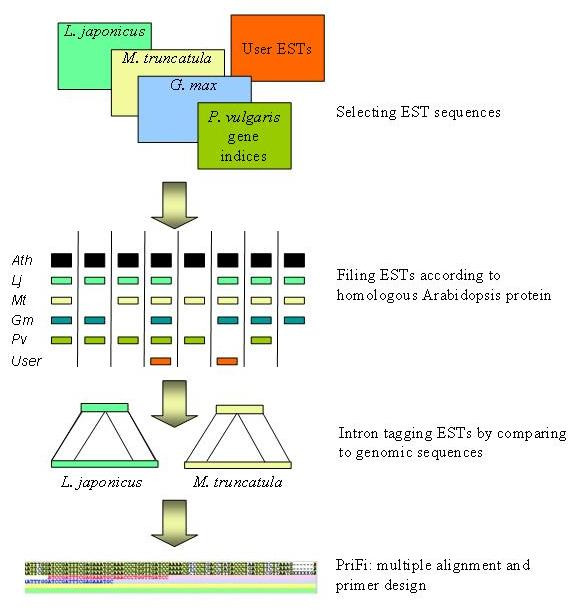
Complete or near-complete genomic sequence information is presently only available for a few plant species representing a large phylogenetic diversity among plants. In order to effectively transfer this information to species lacking sequence information, comparative genomic tools need to be developed. Molecular markers permitting cross-species mapping along co-linear genomic regions are central to comparative genomics. These "anchor" markers, defining unique loci in genetic linkage maps of multiple species, are gene-based and possess a number of features that make them relatively sparse. To identify potential anchor marker sequences more efficiently, we have established an automated bioinformatic pipeline that combines multi-species Expressed Sequence Tags (EST) and genome sequence data.
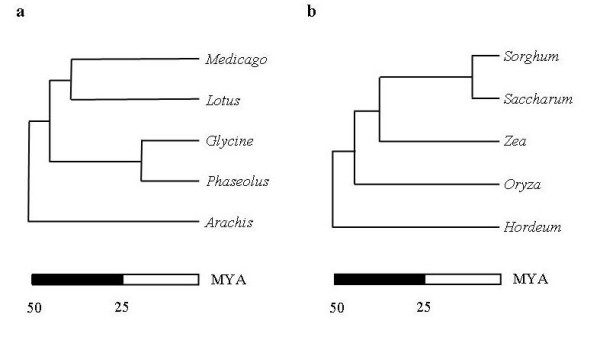
Taking advantage of sequence data from related species, the pipeline identifies evolutionarily conserved sequences that are likely to define unique orthologous loci in most species of the same phylogenetic clade. The key features are the identification of evolutionarily conserved sequences followed by automated design of intron-flanking Polymerase Chain Reaction (PCR) primer pairs. Polymorphisms can subsequently be identified by size- or sequence variation of PCR products, amplified from mapping parents or populations. We illustrate our procedure in legumes and grasses and exemplify its application in legumes, where model plant studies and the genome- and EST-sequence data available have a potential impact on the breeding of crop species and on our understanding of the evolution of this large and diverse family.
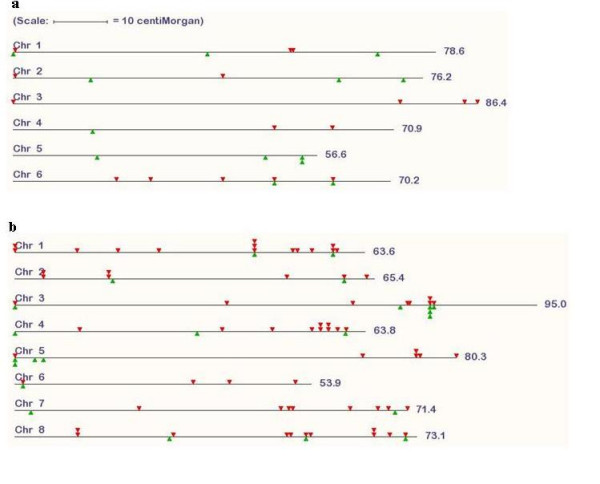
We provide a database of 459 candidate anchor loci which have the potential to serve as map anchors in more than 18,000 legume species, a number of which are of agricultural importance. For grasses, the database contains 1335 candidate anchor loci. Based on this database, we have evaluated 76 candidate anchor loci with respect to marker development in legume species with no sequence information available, demonstrating the validity of this approach.
目前仅少数代表植物中广泛系统发育多样性的植物物种拥有完整或近乎完整的基因组序列信息。为了有效地将这些信息传递到缺乏序列信息的物种,需要开发比较基因组工具。允许跨物种沿共线基因组区域进行定位的分子标记是比较基因组学的核心。这些“锚定”标记在多个物种的遗传连锁图谱中定义了独特的位点,它们基于基因,并且具有一些使其相对稀少的特征。为了更有效地识别潜在的锚定标记序列,我们建立了一个自动化的生物信息学流程,该流程结合了多物种表达序列标签(EST)和基因组序列数据。
利用相关物种的序列数据,该流程识别出可能在同一系统发育分支的大多数物种中定义独特直系同源位点的进化保守序列。关键特征是识别进化保守序列,随后自动设计内含子侧翼聚合酶链反应(PCR)引物对。随后可以通过从定位亲本或群体中扩增的PCR产物的大小或序列变异来识别多态性。我们在豆科植物和禾本科植物中展示了我们的方法,并举例说明了其在豆科植物中的应用,在豆科植物中,模式植物研究以及可用的基因组和EST序列数据可能对作物物种的育种以及我们对这个庞大且多样的科的进化的理解产生影响。
我们提供了一个包含459个候选锚定位点的数据库,这些位点有可能作为超过18000种豆科植物的图谱锚定,其中许多具有农业重要性。对于禾本科植物,该数据库包含1335个候选锚定位点。基于这个数据库,我们针对没有可用序列信息的豆科物种的标记开发评估了76个候选锚定位点,证明了这种方法的有效性。