Jothi Raja, Cherukuri Praveen F, Tasneem Asba, Przytycka Teresa M
National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA.
J Mol Biol. 2006 Sep 29;362(4):861-75. doi: 10.1016/j.jmb.2006.07.072. Epub 2006 Aug 1.
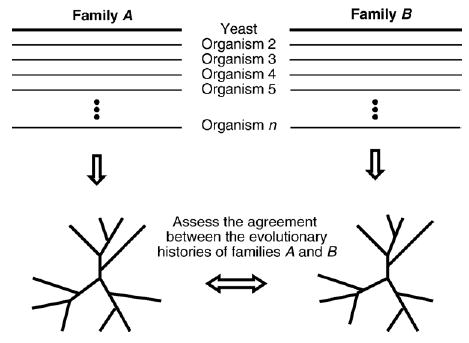
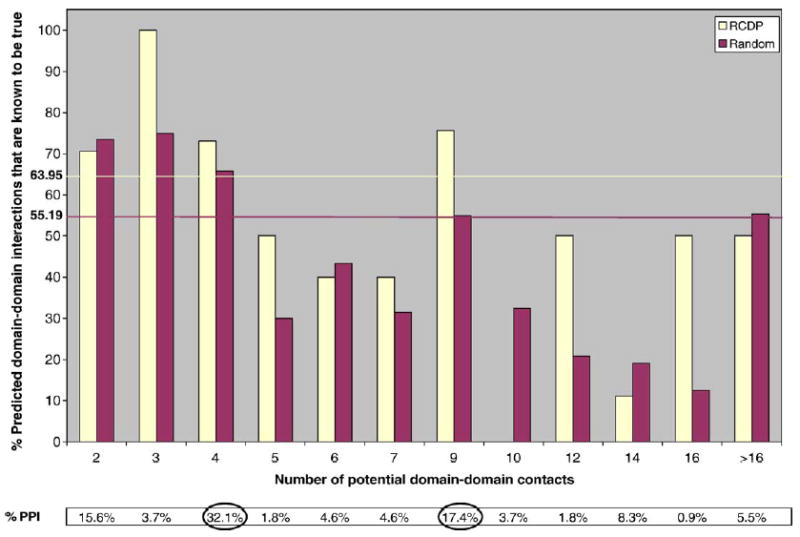
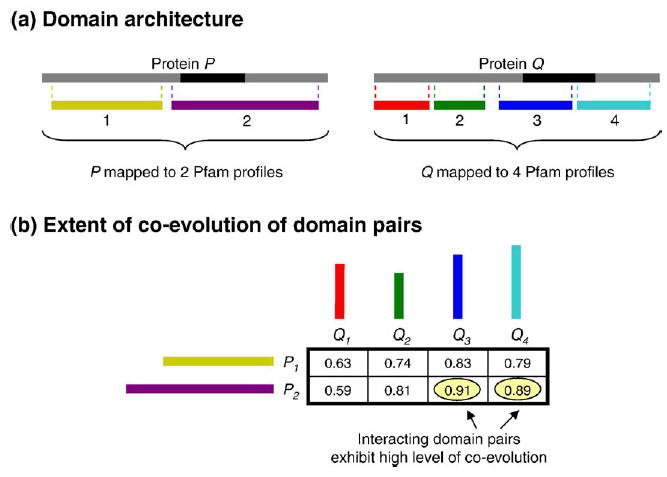
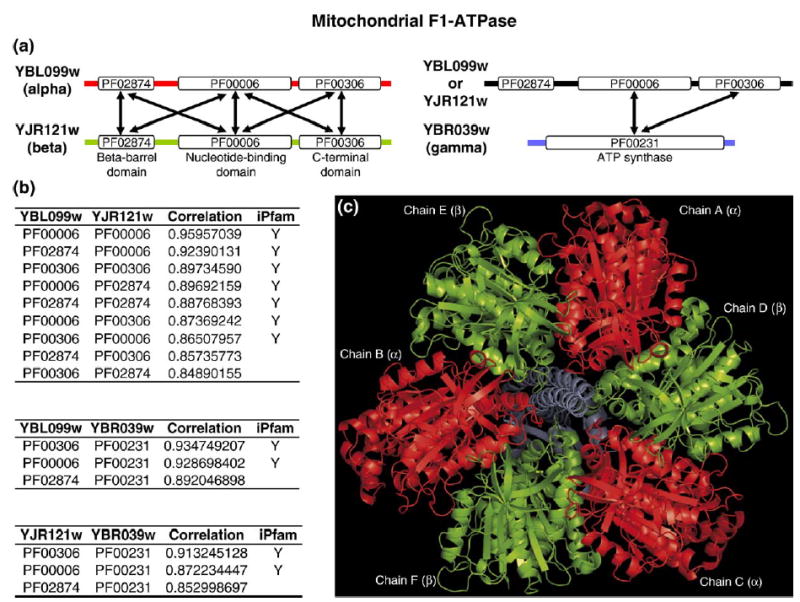
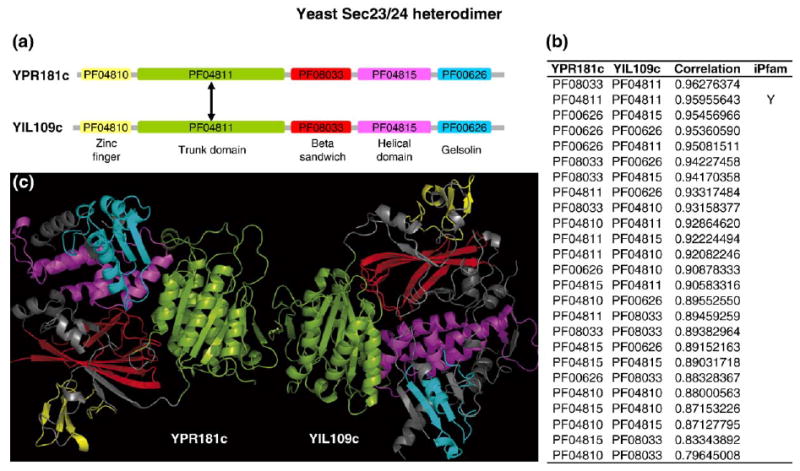
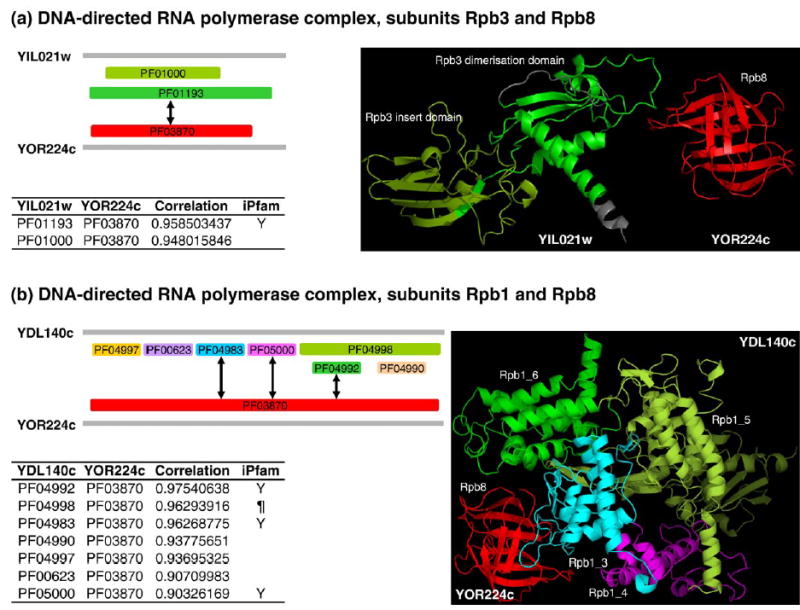
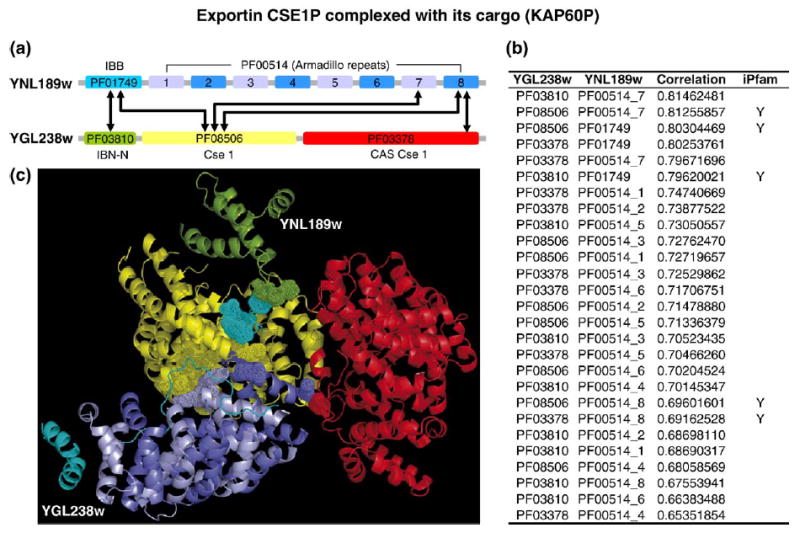
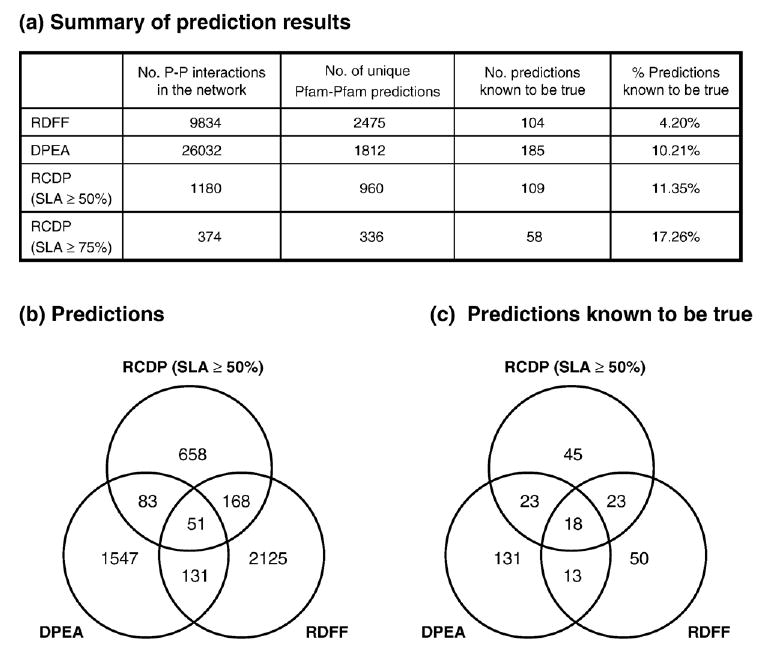
Recent advances in functional genomics have helped generate large-scale high-throughput protein interaction data. Such networks, though extremely valuable towards molecular level understanding of cells, do not provide any direct information about the regions (domains) in the proteins that mediate the interaction. Here, we performed co-evolutionary analysis of domains in interacting proteins in order to understand the degree of co-evolution of interacting and non-interacting domains. Using a combination of sequence and structural analysis, we analyzed protein-protein interactions in F1-ATPase, Sec23p/Sec24p, DNA-directed RNA polymerase and nuclear pore complexes, and found that interacting domain pair(s) for a given interaction exhibits higher level of co-evolution than the non-interacting domain pairs. Motivated by this finding, we developed a computational method to test the generality of the observed trend, and to predict large-scale domain-domain interactions. Given a protein-protein interaction, the proposed method predicts the domain pair(s) that is most likely to mediate the protein interaction. We applied this method on the yeast interactome to predict domain-domain interactions, and used known domain-domain interactions found in PDB crystal structures to validate our predictions. Our results show that the prediction accuracy of the proposed method is statistically significant. Comparison of our prediction results with those from two other methods reveals that only a fraction of predictions are shared by all the three methods, indicating that the proposed method can detect known interactions missed by other methods. We believe that the proposed method can be used with other methods to help identify previously unrecognized domain-domain interactions on a genome scale, and could potentially help reduce the search space for identifying interaction sites.
功能基因组学的最新进展有助于生成大规模高通量蛋白质相互作用数据。这类网络虽然对于从分子水平理解细胞极有价值,但并未提供有关介导相互作用的蛋白质区域(结构域)的任何直接信息。在此,我们对相互作用蛋白质中的结构域进行了共进化分析,以了解相互作用和非相互作用结构域的共进化程度。通过结合序列和结构分析,我们分析了F1 - ATP酶、Sec23p/Sec24p、DNA指导的RNA聚合酶和核孔复合体中的蛋白质 - 蛋白质相互作用,发现给定相互作用的相互作用结构域对表现出比非相互作用结构域对更高水平的共进化。受这一发现的启发,我们开发了一种计算方法来测试观察到的趋势的普遍性,并预测大规模的结构域 - 结构域相互作用。给定一个蛋白质 - 蛋白质相互作用,该方法预测最有可能介导蛋白质相互作用的结构域对。我们将此方法应用于酵母相互作用组以预测结构域 - 结构域相互作用,并使用在PDB晶体结构中发现的已知结构域 - 结构域相互作用来验证我们的预测。我们的结果表明,该方法的预测准确性具有统计学意义。将我们的预测结果与其他两种方法的结果进行比较发现,所有三种方法仅共享一小部分预测结果,这表明该方法可以检测到其他方法遗漏的已知相互作用。我们相信,该方法可与其他方法一起用于在基因组规模上帮助识别以前未被认识的结构域 - 结构域相互作用,并可能有助于缩小识别相互作用位点的搜索空间。