Rapin Isabelle, Weidenheim Karen, Lindenbaum Yelena, Rosenbaum Pearl, Merchant Saumil N, Krishna Sindu, Dickson Dennis W
Albert Einstein College of Medicine, Bronx, NY 10461, USA.
J Child Neurol. 2006 Nov;21(11):991-1006. doi: 10.1177/08830738060210110101.
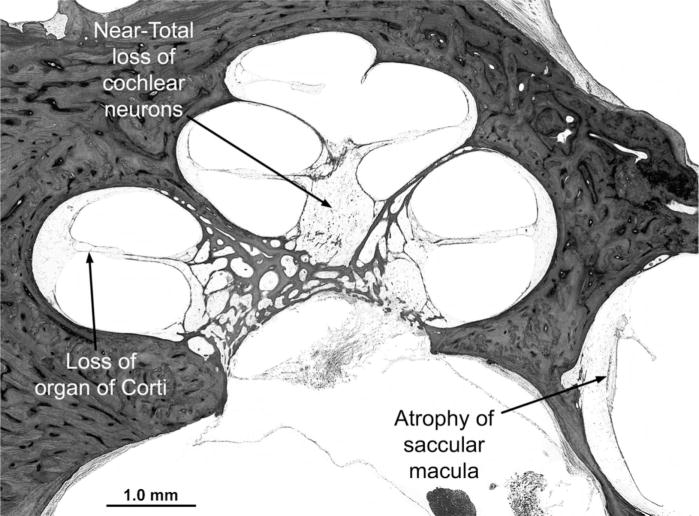
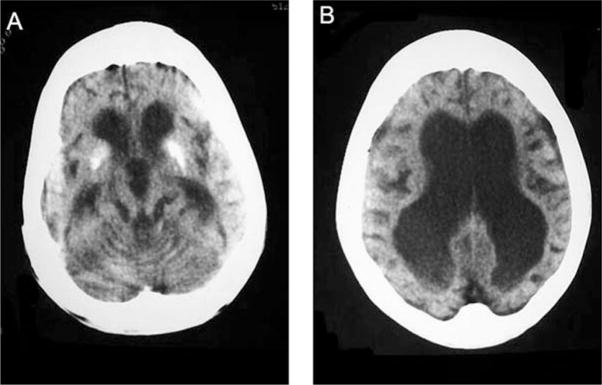
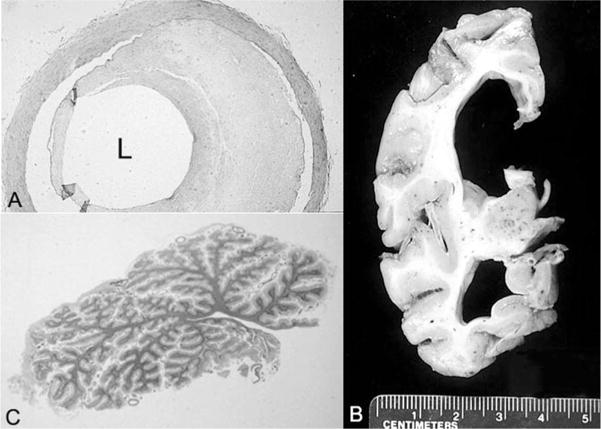
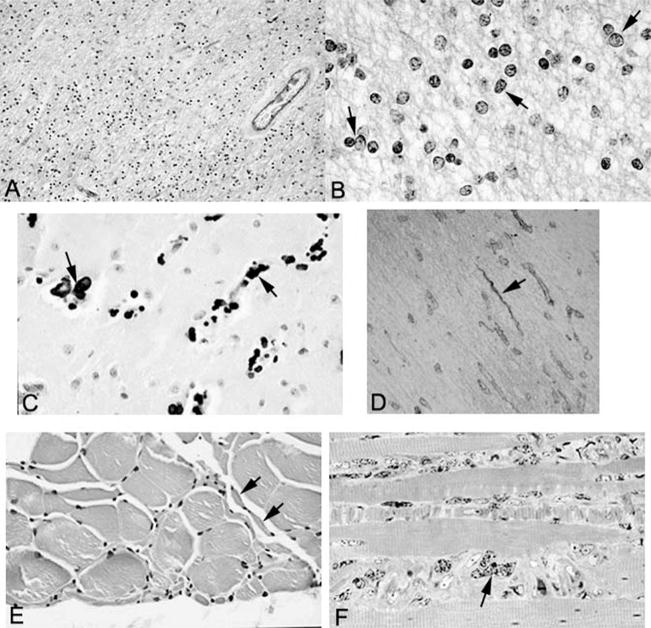
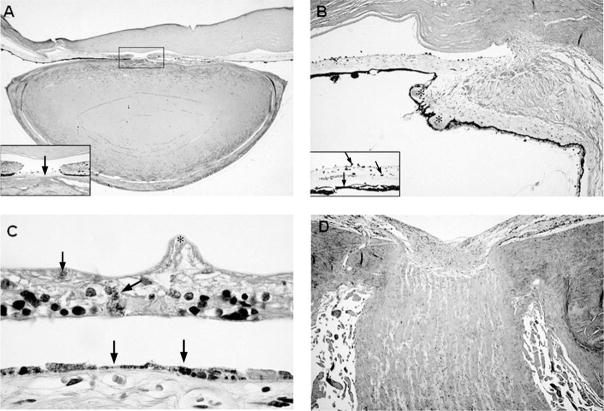
Cockayne syndrome and xeroderma pigmentosum-Cockayne syndrome complex are rare autosomal recessive disorders with poorly understood biology. They are characterized by profound postnatal brain and somatic growth failure and by degeneration of multiple tissues resulting in cachexia, dementia, and premature aging. They result in premature death, usually in childhood, exceptionally in adults. This study compares the clinical course and pathology of a man with Cockayne syndrome group A who died at age 31(1/2) years with 15 adequately documented other adults with Cockayne syndrome and 5 with xeroderma pigmentosum-Cockayne syndrome complex. Slowing of head and somatic growth was apparent before age 2 years, mental retardation and slowly progressive spasticity at 4 years, ataxia and hearing loss at 9 years, visual impairment at 14 years, typical Cockayne facies at 17 years, and cachexia and dementia in his twenties, with a retained outgoing personality. He experienced several transient right and left hemipareses and two episodes of status epilepticus following falls. Neuropathology disclosed profound microencephaly, bilateral old subdural hematomas, white-matter atrophy, tigroid leukodystrophy with string vessels, oligodendrocyte proliferation, bizarre reactive astrocytes, multifocal dystrophic calcification that was most marked in the basal ganglia, advanced atherosclerosis, mixed demyelinating and axonal neuropathy, and neurogenic muscular atrophy. Cellular degeneration of the organ of Corti, spiral and vestibular ganglia, and all chambers of the eye was severe. Rarely, and for unexplained reasons, in some patients with Cockayne syndrome the course is slower than usual, resulting in survival into adulthood. The profound dwarfing, failure of brain growth, cachexia, selectivity of tissue degeneration, and poor correlation between genotypes and phenotypes are not understood. Deficient repair of DNA can increase vulnerability to oxidative stress and play a role in the premature aging, but why patients with mutations in xeroderma pigmentosum genes present with the Cockayne syndrome phenotype is still not known.
科凯恩综合征和着色性干皮病 - 科凯恩综合征复合体是罕见的常染色体隐性疾病,其生物学机制尚不清楚。它们的特征是出生后脑和身体生长严重迟缓,以及多个组织的退化,导致恶病质、痴呆和早衰。它们会导致过早死亡,通常在儿童期,极少数情况下在成年期。本研究比较了一名31岁半死于A组科凯恩综合征男性的临床病程和病理情况,与另外15名有充分记录的成年科凯恩综合征患者以及5名着色性干皮病 - 科凯恩综合征复合体患者。2岁前头部和身体生长明显减缓,4岁时出现智力迟钝和缓慢进展的痉挛,9岁时出现共济失调和听力丧失,14岁时出现视力障碍,17岁时出现典型的科凯恩面容,二十多岁时出现恶病质和痴呆,但其外向性格得以保留。他经历了几次短暂的左右偏瘫,跌倒后出现两次癫痫持续状态。神经病理学显示严重的小头畸形、双侧陈旧性硬膜下血肿、白质萎缩、伴有条索状血管的虎斑状脑白质营养不良、少突胶质细胞增殖、怪异的反应性星形胶质细胞、多灶性营养不良性钙化(在基底神经节最为明显)、晚期动脉粥样硬化、混合性脱髓鞘和轴索性神经病变以及神经源性肌肉萎缩。柯蒂氏器、螺旋神经节和前庭神经节以及眼的所有腔室的细胞退化严重。在某些科凯恩综合征患者中,罕见且原因不明的是,病程比通常情况要慢,从而存活至成年。严重的侏儒症、脑生长发育不良、恶病质、组织退化的选择性以及基因型与表型之间的低相关性尚不清楚。DNA修复缺陷会增加对氧化应激的易感性,并在早衰中起作用,但为什么着色性干皮病基因突变的患者会表现出科凯恩综合征的表型仍然未知。