Cardiff School of Biosciences, Cardiff University, United Kingdom.
PLoS One. 2006 Dec 20;1(1):e17. doi: 10.1371/journal.pone.0000017.
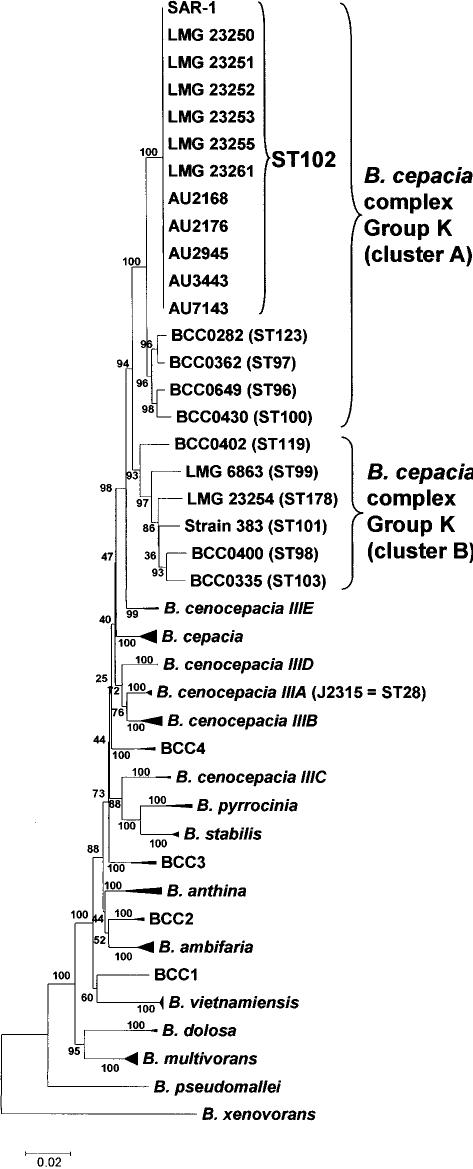
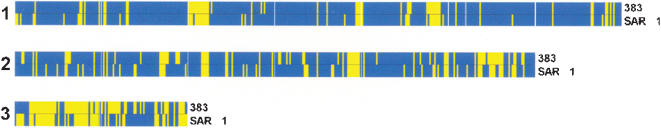
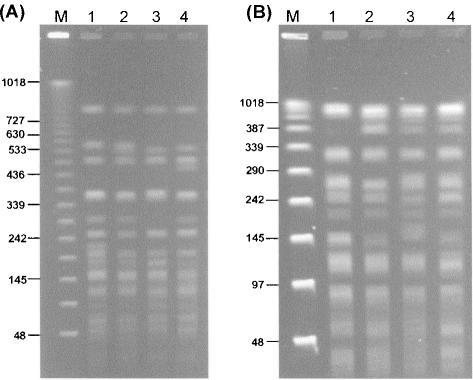
Shot-gun sequencing of DNA isolated from the environment and the assembly of metagenomes from the resulting data has considerably advanced the study of microbial diversity. However, the subsequent matching of these hypothetical metagenomes to cultivable microorganisms is a limitation of such cultivation-independent methods of population analysis. Using a nucleotide sequence-based genetic typing method, multilocus sequence typing, we were able for the first time to match clonal cultivable isolates to a published and controversial bacterial metagenome, Burkholderia SAR-1, which derived from analysis of the Sargasso Sea. The matching cultivable isolates were all associated with infection and geographically widely distributed; taxonomic analysis demonstrated they were members of Burkholderia cepacia complex Group K. Comparison of the Burkholderia SAR-1 metagenome to closely related B. cepacia complex genomes indicated that it was greater than 98% intact in terms of conserved genes, and it also shared complete sequence identity with the cultivable isolates at random loci beyond the genes sampled by the multilocus sequence typing. Two features of the extant cultivable clones support the argument that the Burkholderia SAR-1 sequence may have been a contaminant in the original metagenomic survey: (i) their growth in conditions reflective of sea water was poor, suggesting the ocean was not their preferred habitat, and (ii) several of the matching isolates were epidemiologically linked to outbreaks of infection that resulted from contaminated medical devices or products, indicating an adaptive fitness of this bacterial strain towards contamination-associated environments. The ability to match identical cultivable strains of bacteria to a hypothetical metagenome is a unique feature of nucleotide sequence-based microbial typing methods; such matching would not have been possible with more traditional methods of genetic typing, such as those based on pattern matching of genomic restriction fragments or amplified DNA fragments. Overall, we have taken the first steps in moving the status of the Burkholderia SAR-1 metagenome from a hypothetical entity towards the basis for life of cultivable strains that may now be analysed in conjunction with the assembled metagenomic sequence data by the wider scientific community.
从环境中分离出的 DNA 进行散弹枪测序,并对由此产生的数据进行宏基因组组装,这极大地促进了微生物多样性的研究。然而,将这些假设的宏基因组与可培养微生物相匹配是这种依赖培养的种群分析方法的一个局限性。我们使用基于核苷酸序列的遗传分型方法——多位点序列分型,首次能够将克隆可培养分离株与已发表的、有争议的细菌宏基因组 Burkholderia SAR-1 相匹配,该宏基因组源自马尾藻海的分析。可匹配的可培养分离株均与感染有关,且分布广泛;分类分析表明,它们是伯克霍尔德氏菌复合群 K 的成员。将 Burkholderia SAR-1 宏基因组与密切相关的伯克霍尔德氏菌复合群基因组进行比较表明,就保守基因而言,它的完整性超过 98%,并且在通过多位点序列分型采样的基因之外的随机基因座上与可培养分离株具有完全的序列同一性。现有的可培养克隆体的两个特征支持这样的论点,即 Burkholderia SAR-1 序列可能是原始宏基因组调查中的污染物:(i) 它们在反映海水条件下的生长情况较差,这表明海洋不是它们的首选栖息地,以及 (ii) 几个匹配的分离株与受污染的医疗器械或产品引起的感染爆发有关,这表明该细菌菌株对污染相关环境具有适应性。将相同的可培养细菌菌株与假设的宏基因组相匹配是基于核苷酸序列的微生物分型方法的独特特征;使用传统的遗传分型方法(例如基于基因组限制性片段或扩增 DNA 片段模式匹配的方法)是不可能进行这种匹配的。总体而言,我们已经迈出了第一步,使 Burkholderia SAR-1 宏基因组从一个假设实体的状态转变为可培养菌株的生命基础,现在更广泛的科学界可以结合组装的宏基因组序列数据对其进行分析。