Redelings Benjamin D, Suchard Marc A
Bioinformatics Research Center, North Carolina State University, Raleigh, NC 27606, USA.
BMC Evol Biol. 2007 Mar 14;7:40. doi: 10.1186/1471-2148-7-40.
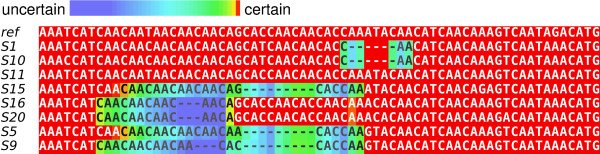
Phylogenies of rapidly evolving pathogens can be difficult to resolve because of the small number of substitutions that accumulate in the short times since divergence. To improve resolution of such phylogenies we propose using insertion and deletion (indel) information in addition to substitution information. We accomplish this through joint estimation of alignment and phylogeny in a Bayesian framework, drawing inference using Markov chain Monte Carlo. Joint estimation of alignment and phylogeny sidesteps biases that stem from conditioning on a single alignment by taking into account the ensemble of near-optimal alignments.
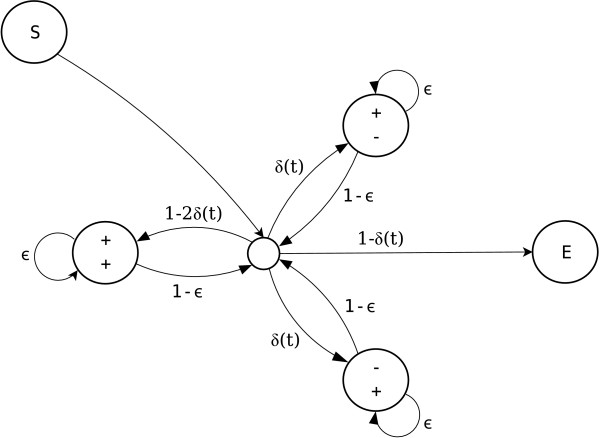
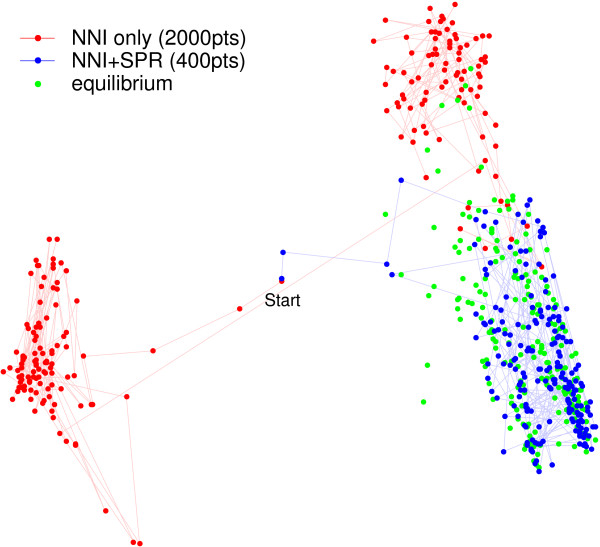
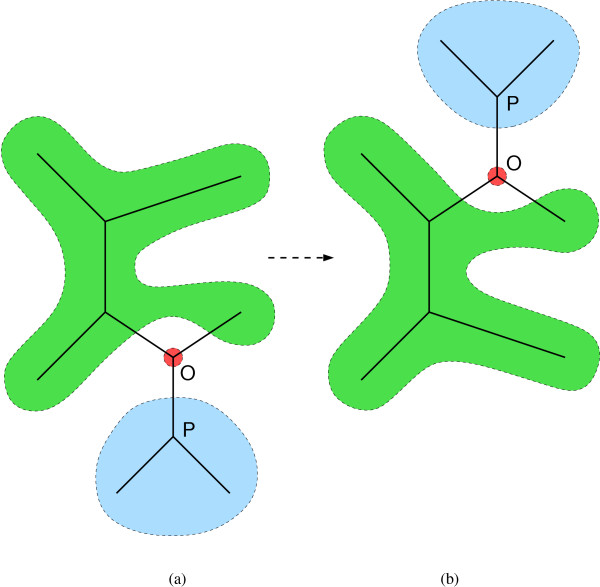
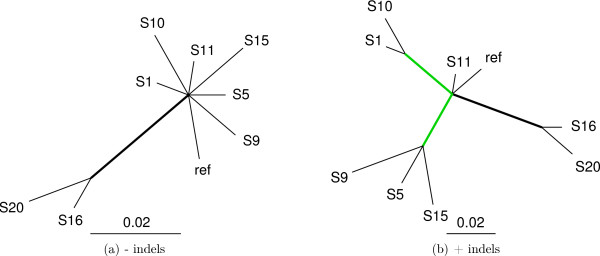
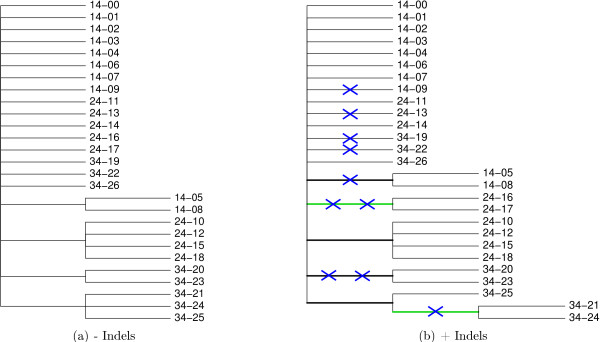
We introduce a novel Markov chain transition kernel that improves computational efficiency by proposing non-local topology rearrangements and by block sampling alignment and topology parameters. In addition, we extend our previous indel model to increase biological realism by placing indels preferentially on longer branches. We demonstrate the ability of indel information to increase phylogenetic resolution in examples drawn from within-host viral sequence samples. We also demonstrate the importance of taking alignment uncertainty into account when using such information. Finally, we show that codon-based substitution models can significantly affect alignment quality and phylogenetic inference by unrealistically forcing indels to begin and end between codons.
These results indicate that indel information can improve phylogenetic resolution of recently diverged pathogens and that alignment uncertainty should be considered in such analyses.
由于快速进化的病原体自分化以来在短时间内积累的替换数量较少,其系统发育关系可能难以解析。为了提高此类系统发育关系的解析度,我们建议除了使用替换信息外,还使用插入和缺失(indel)信息。我们通过在贝叶斯框架中对序列比对和系统发育进行联合估计来实现这一点,并使用马尔可夫链蒙特卡罗进行推断。序列比对和系统发育的联合估计通过考虑近乎最优比对的集合,避免了因基于单个比对而产生的偏差。
我们引入了一种新颖的马尔可夫链转移核,通过提出非局部拓扑重排以及对序列比对和拓扑参数进行分块采样来提高计算效率。此外,我们扩展了之前的indel模型,通过将indel优先放置在较长分支上来增加生物学真实性。我们在宿主内病毒序列样本的实例中展示了indel信息增加系统发育解析度的能力。我们还证明了在使用此类信息时考虑序列比对不确定性的重要性。最后,我们表明基于密码子的替换模型可能会通过不切实际地迫使indel在密码子之间开始和结束,从而显著影响序列比对质量和系统发育推断。
这些结果表明,indel信息可以提高近期分化病原体的系统发育解析度,并且在这类分析中应考虑序列比对的不确定性。