Bernal Axel, Crammer Koby, Hatzigeorgiou Artemis, Pereira Fernando
Department of Computer and Information Science, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America.
PLoS Comput Biol. 2007 Mar 16;3(3):e54. doi: 10.1371/journal.pcbi.0030054. Epub 2007 Feb 2.
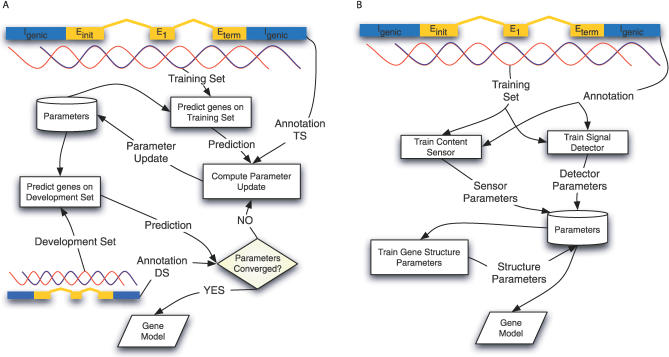
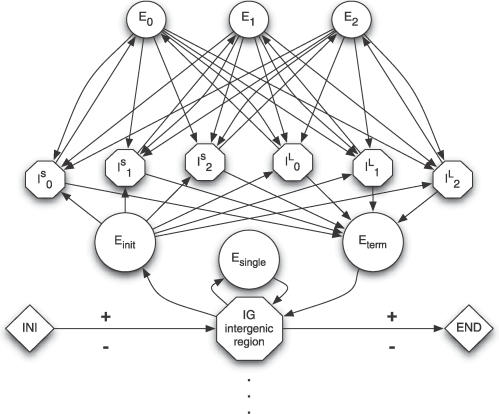
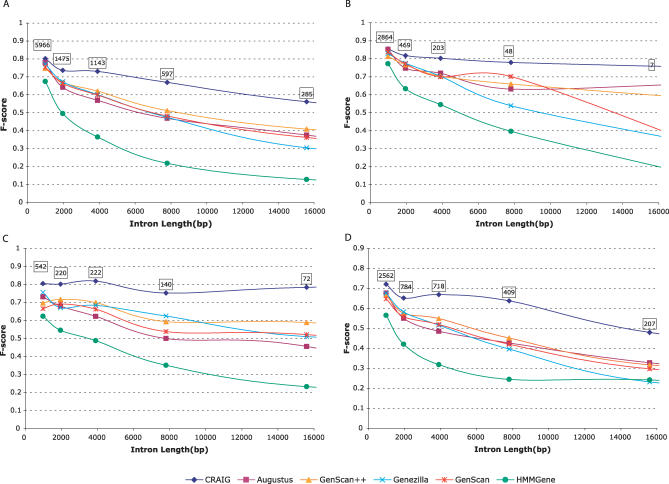
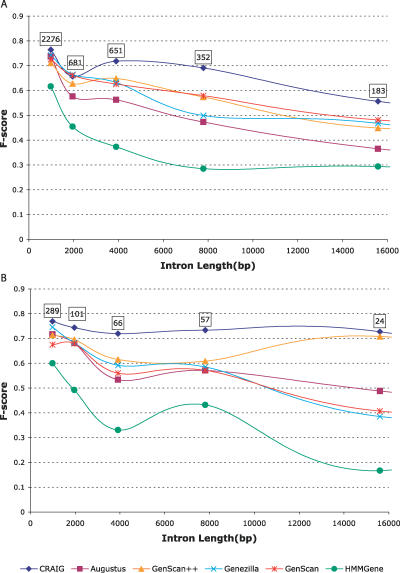
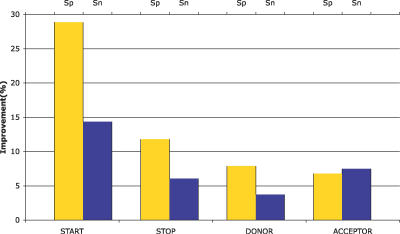
Most ab initio gene predictors use a probabilistic sequence model, typically a hidden Markov model, to combine separately trained models of genomic signals and content. By combining separate models of relevant genomic features, such gene predictors can exploit small training sets and incomplete annotations, and can be trained fairly efficiently. However, that type of piecewise training does not optimize prediction accuracy and has difficulty in accounting for statistical dependencies among different parts of the gene model. With genomic information being created at an ever-increasing rate, it is worth investigating alternative approaches in which many different types of genomic evidence, with complex statistical dependencies, can be integrated by discriminative learning to maximize annotation accuracy. Among discriminative learning methods, large-margin classifiers have become prominent because of the success of support vector machines (SVM) in many classification tasks. We describe CRAIG, a new program for ab initio gene prediction based on a conditional random field model with semi-Markov structure that is trained with an online large-margin algorithm related to multiclass SVMs. Our experiments on benchmark vertebrate datasets and on regions from the ENCODE project show significant improvements in prediction accuracy over published gene predictors that use intrinsic features only, particularly at the gene level and on genes with long introns.
大多数从头开始的基因预测器使用概率序列模型,通常是隐马尔可夫模型,来组合分别训练的基因组信号和内容模型。通过组合相关基因组特征的单独模型,此类基因预测器可以利用小训练集和不完整注释,并且可以相当高效地进行训练。然而,那种类型的分段训练并不能优化预测准确性,并且难以考虑基因模型不同部分之间的统计依赖性。随着基因组信息以不断增加的速度产生,值得研究替代方法,在这些方法中,可以通过判别式学习整合具有复杂统计依赖性的许多不同类型的基因组证据,以最大化注释准确性。在判别式学习方法中,大间隔分类器因其在许多分类任务中支持向量机(SVM)的成功而变得突出。我们描述了CRAIG,这是一个基于具有半马尔可夫结构的条件随机场模型的从头开始基因预测新程序,该模型使用与多类SVM相关的在线大间隔算法进行训练。我们在基准脊椎动物数据集和ENCODE项目区域上的实验表明,与仅使用内在特征的已发表基因预测器相比,预测准确性有显著提高,特别是在基因水平以及具有长内含子的基因上。