Desai Dhwani K, Nandi Soumyadeep, Srivastava Prashant K, Lynn Andrew M
Biological Oceanography Division, Leibniz Institute of Marine Sciences (IFM-GEOMAR), Düsternbrooker Weg 20, 24105 Kiel, Germany.
Adv Bioinformatics. 2011;2011:743782. doi: 10.1155/2011/743782. Epub 2011 Mar 29.
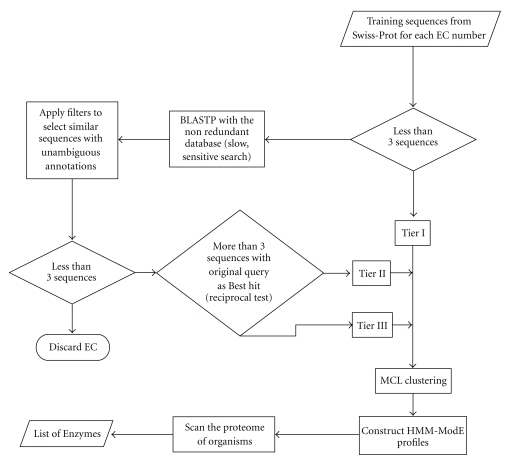
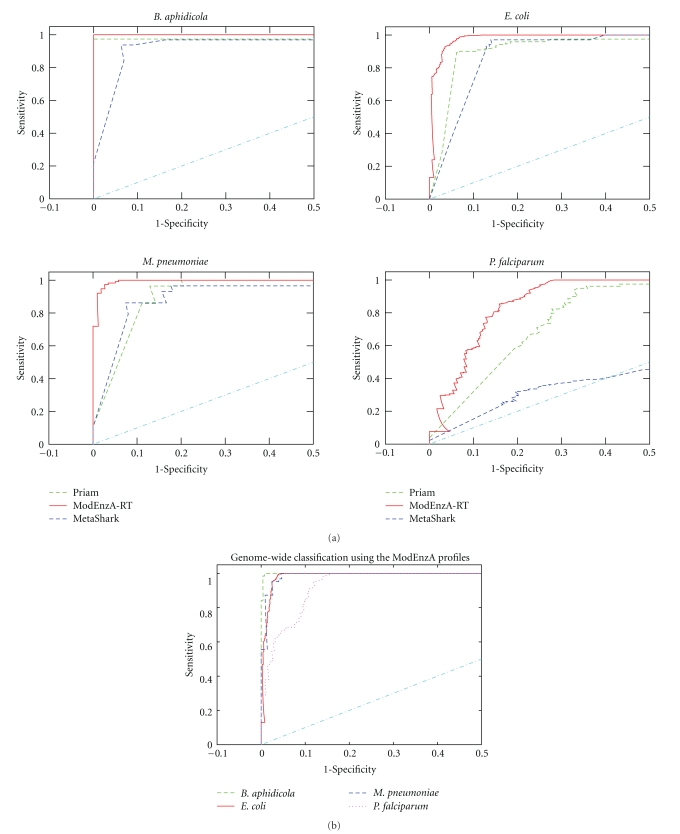
Various enzyme identification protocols involving homology transfer by sequence-sequence or profile-sequence comparisons have been devised which utilise Swiss-Prot sequences associated with EC numbers as the training set. A profile HMM constructed for a particular EC number might select sequences which perform a different enzymatic function due to the presence of certain fold-specific residues which are conserved in enzymes sharing a common fold. We describe a protocol, ModEnzA (HMM-ModE Enzyme Annotation), which generates profile HMMs highly specific at a functional level as defined by the EC numbers by incorporating information from negative training sequences. We enrich the training dataset by mining sequences from the NCBI Non-Redundant database for increased sensitivity. We compare our method with other enzyme identification methods, both for assigning EC numbers to a genome as well as identifying protein sequences associated with an enzymatic activity. We report a sensitivity of 88% and specificity of 95% in identifying EC numbers and annotating enzymatic sequences from the E. coli genome which is higher than any other method. With the next-generation sequencing methods producing a huge amount of sequence data, the development and use of fully automated yet accurate protocols such as ModEnzA is warranted for rapid annotation of newly sequenced genomes and metagenomic sequences.
已经设计了各种酶识别方案,这些方案通过序列-序列或轮廓-序列比较进行同源性转移,利用与酶委员会(EC)编号相关的瑞士蛋白质数据库(Swiss-Prot)序列作为训练集。为特定EC编号构建的轮廓隐马尔可夫模型(profile HMM)可能会选择由于存在某些在具有共同折叠的酶中保守的折叠特异性残基而执行不同酶功能的序列。我们描述了一种方案,即ModEnzA(HMM-ModE酶注释),它通过纳入来自阴性训练序列的信息,生成在EC编号定义的功能水平上具有高度特异性的轮廓HMM。我们通过从NCBI非冗余数据库挖掘序列来丰富训练数据集,以提高灵敏度。我们将我们的方法与其他酶识别方法进行比较,包括为基因组分配EC编号以及识别与酶活性相关的蛋白质序列。我们报告在识别EC编号和注释大肠杆菌基因组中的酶序列时,灵敏度为88%,特异性为95%,高于任何其他方法。随着下一代测序方法产生大量序列数据,像ModEnzA这样的全自动且准确的方案的开发和使用对于新测序基因组和宏基因组序列的快速注释是必要的。