Tjong Harianto, Qin Sanbo, Zhou Huan-Xiang
Institute of Molecular Biophysics and School of Computational Science and Department of Physics, Florida State University, Tallahassee, FL 32306, USA.
Nucleic Acids Res. 2007 Jul;35(Web Server issue):W357-62. doi: 10.1093/nar/gkm231. Epub 2007 May 25.
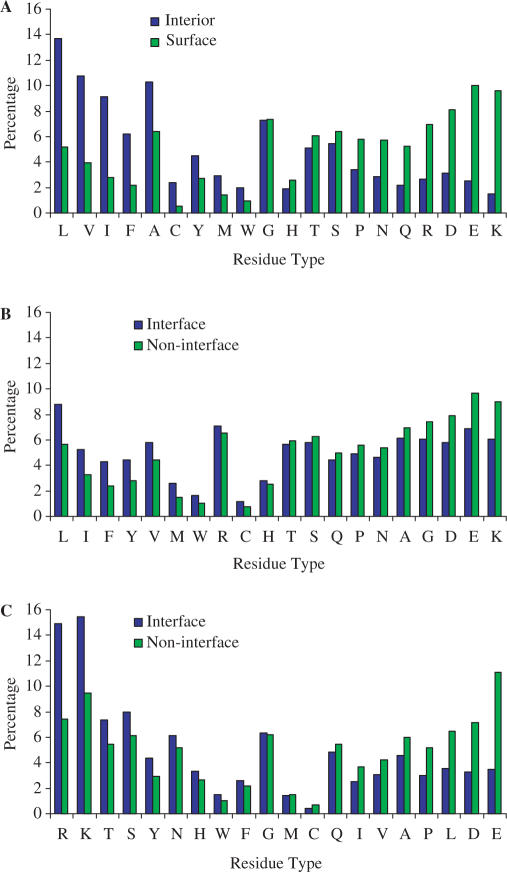
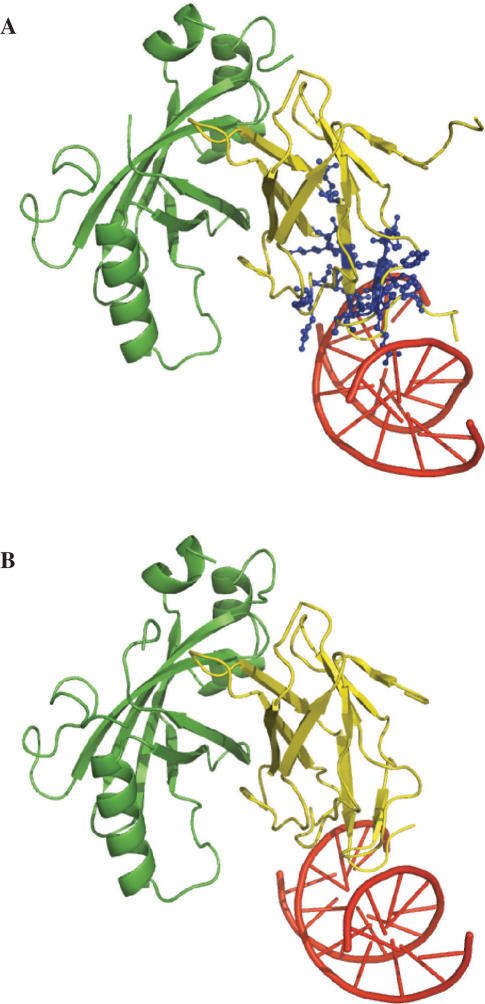
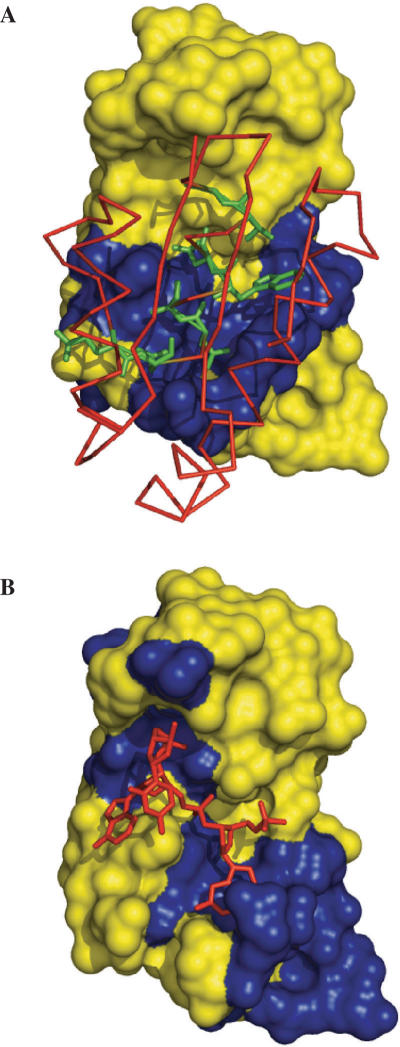
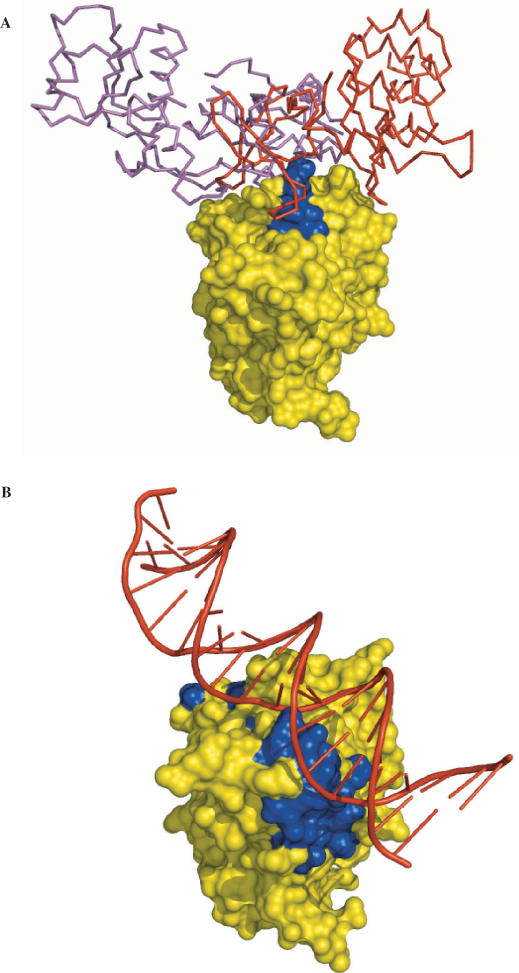
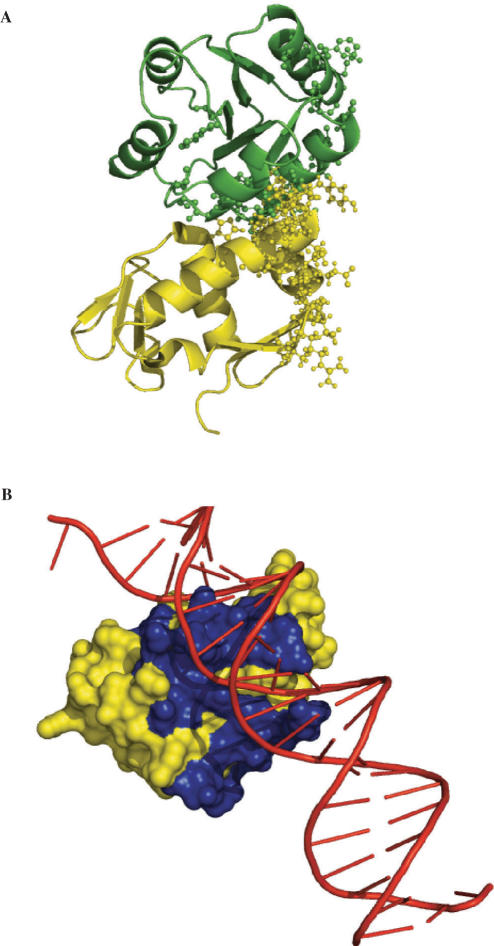
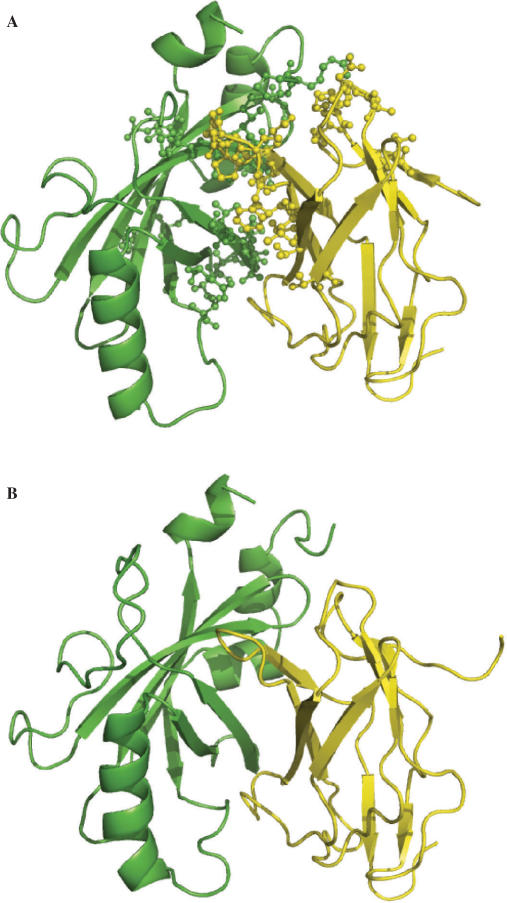
The side chains of the 20 types of amino acids, owing to a large extent to their different physical properties, have characteristic distributions in interior/surface regions of individual proteins and in interface/non-interface portions of protein surfaces that bind proteins or nucleic acids. These distributions have important structural and functional implications. We have developed accurate methods for predicting the solvent accessibility of amino acids from a protein sequence and for predicting interface residues from the structure of a protein-binding or DNA-binding protein. The methods are called WESA, cons-PPISP and DISPLAR, respectively. The web servers of these methods are now available at http://pipe.scs.fsu.edu. To illustrate the utility of these web servers, cons-PPISP and DISPLAR predictions are used to construct a structural model for a multicomponent protein-DNA complex.
20种氨基酸的侧链在很大程度上由于其不同的物理性质,在单个蛋白质的内部/表面区域以及结合蛋白质或核酸的蛋白质表面的界面/非界面部分具有特征性分布。这些分布具有重要的结构和功能意义。我们已经开发出了从蛋白质序列预测氨基酸溶剂可及性以及从蛋白质结合或DNA结合蛋白的结构预测界面残基的准确方法。这些方法分别称为WESA、cons-PPISP和DISPLAR。这些方法的网络服务器现在可在http://pipe.scs.fsu.edu上获取。为了说明这些网络服务器的实用性,使用cons-PPISP和DISPLAR预测来构建多组分蛋白质-DNA复合物的结构模型。