Hogg Justin S, Hu Fen Z, Janto Benjamin, Boissy Robert, Hayes Jay, Keefe Randy, Post J Christopher, Ehrlich Garth D
Allegheny General Hospital, Allegheny-Singer Research Institute, Center for Genomic Sciences, Pittsburgh, Pennsylvania 15212, USA.
Genome Biol. 2007;8(6):R103. doi: 10.1186/gb-2007-8-6-r103.
The distributed genome hypothesis (DGH) posits that chronic bacterial pathogens utilize polyclonal infection and reassortment of genic characters to ensure persistence in the face of adaptive host defenses. Studies based on random sequencing of multiple strain libraries suggested that free-living bacterial species possess a supragenome that is much larger than the genome of any single bacterium.
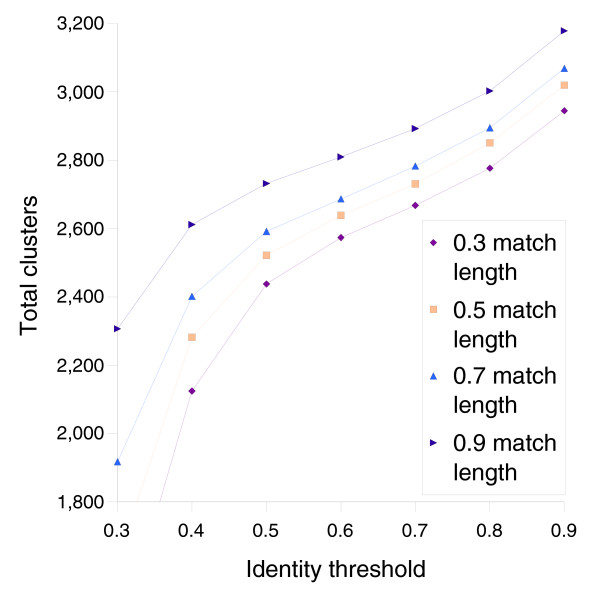
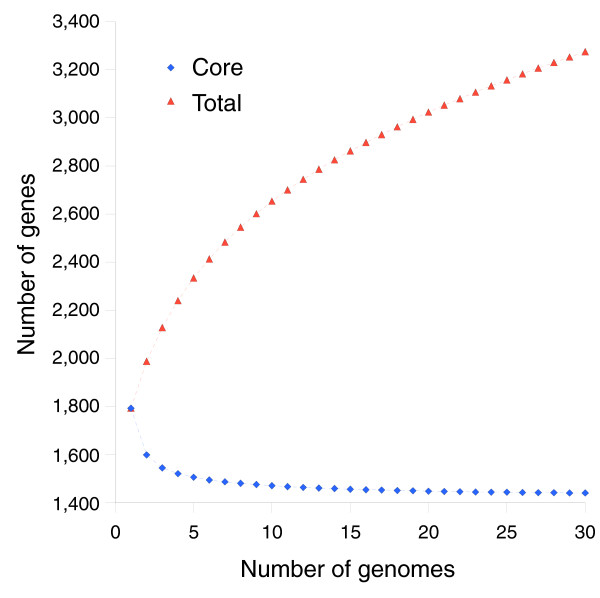
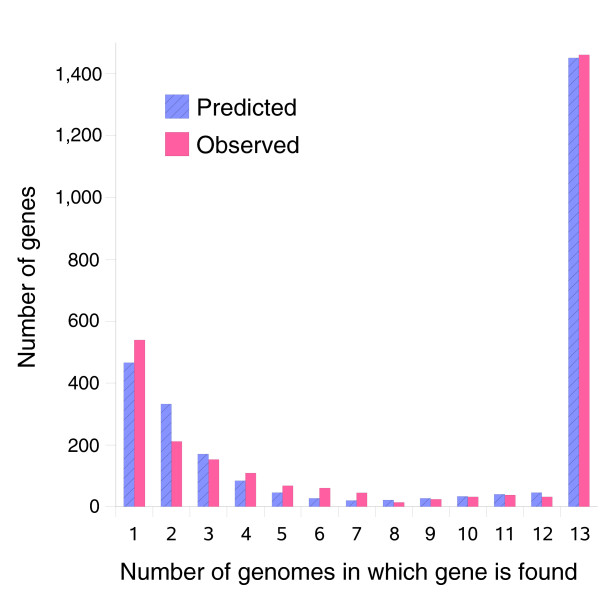
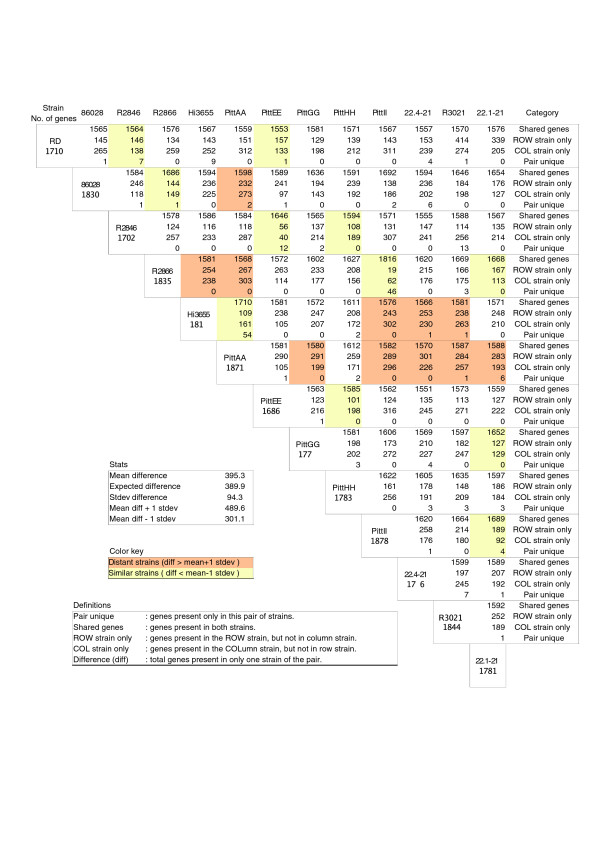
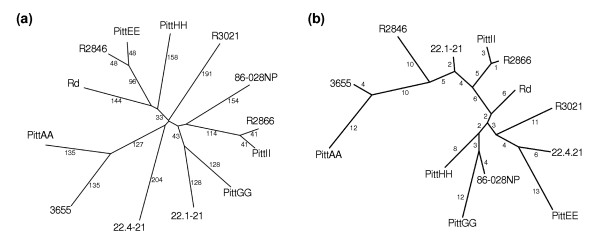
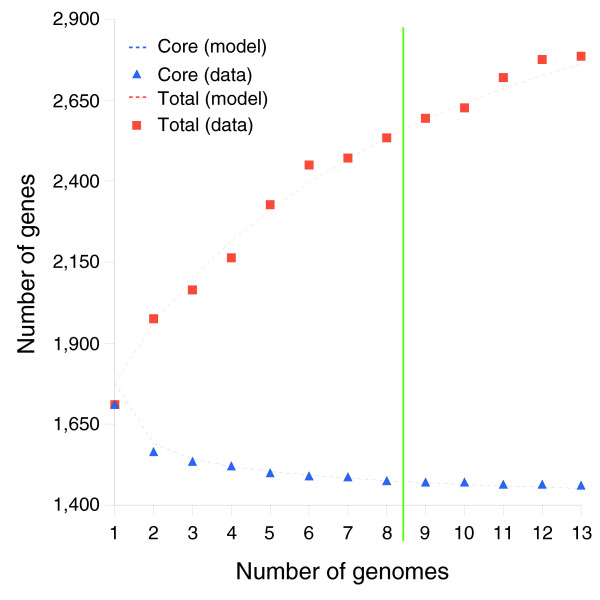
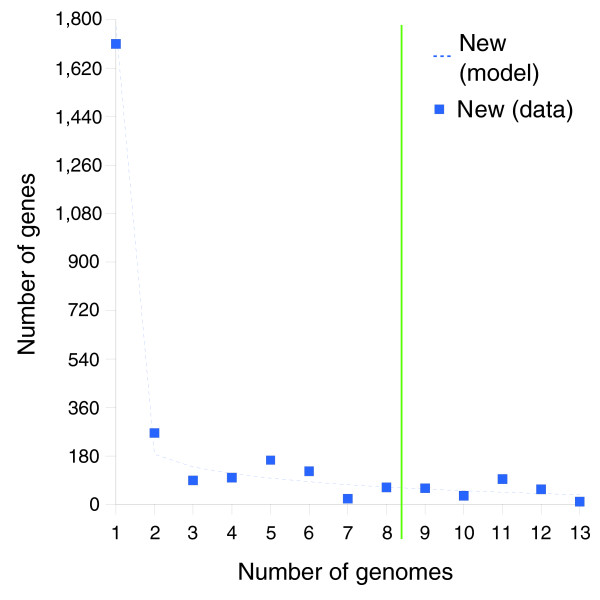
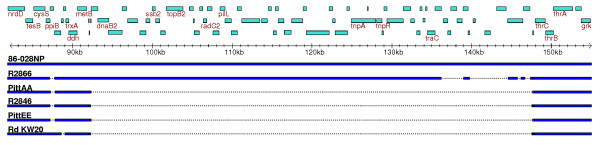
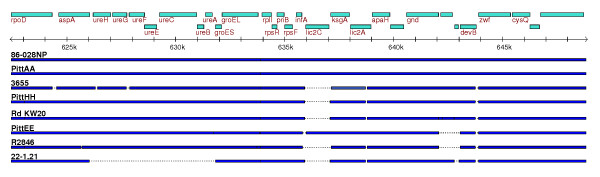
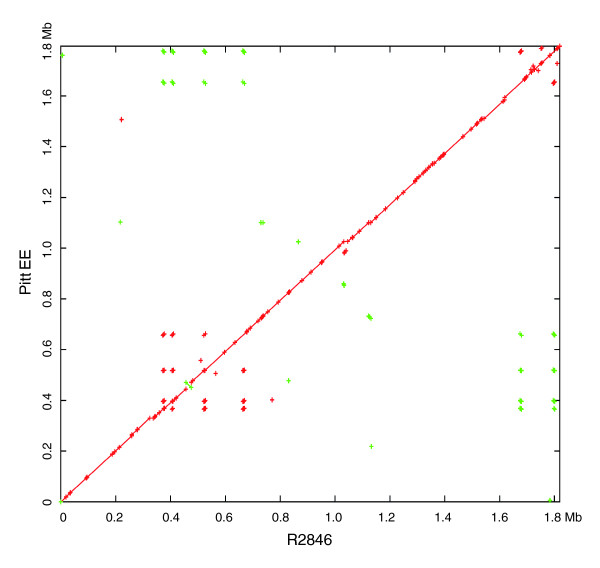
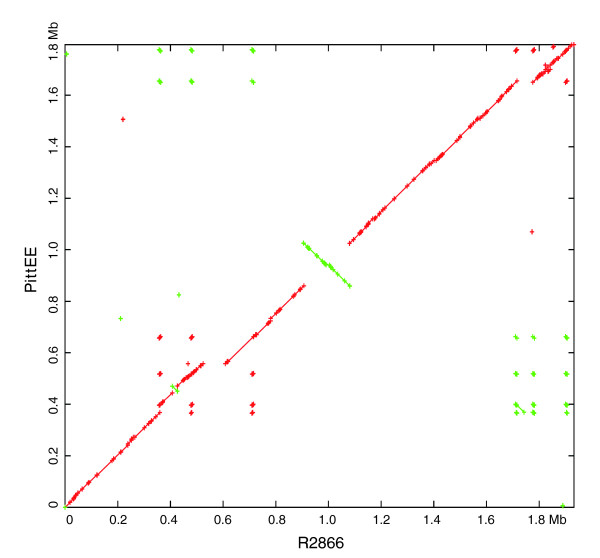
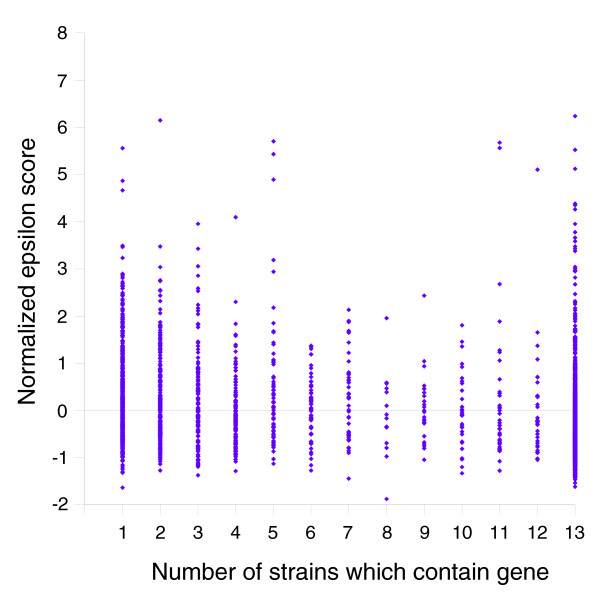
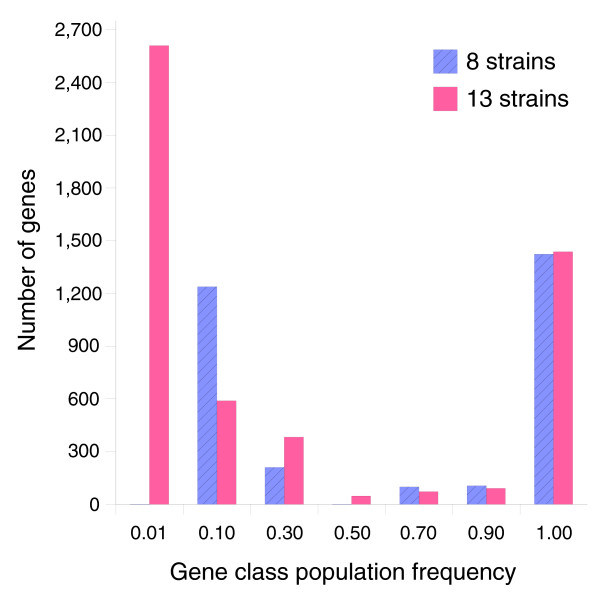
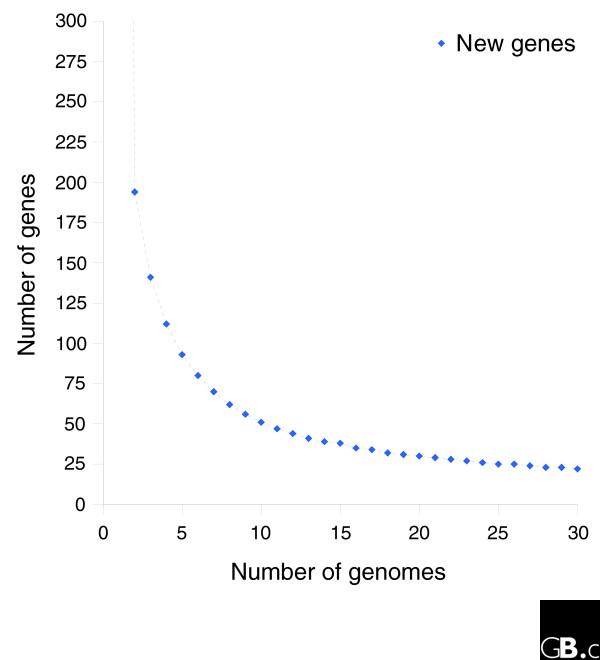
We derived high depth genomic coverage of nine nontypeable Haemophilus influenzae (NTHi) clinical isolates, bringing to 13 the number of sequenced NTHi genomes. Clustering identified 2,786 genes, of which 1,461 were common to all strains, with each of the remaining 1,328 found in a subset of strains; the number of clusters ranged from 1,686 to 1,878 per strain. Genic differences of between 96 and 585 were identified per strain pair. Comparisons of each of the NTHi strains with the Rd strain revealed between 107 and 158 insertions and 100 and 213 deletions per genome. The mean insertion and deletion sizes were 1,356 and 1,020 base-pairs, respectively, with mean maximum insertions and deletions of 26,977 and 37,299 base-pairs. This relatively large number of small rearrangements among strains is in keeping with what is known about the transformation mechanisms in this naturally competent pathogen.
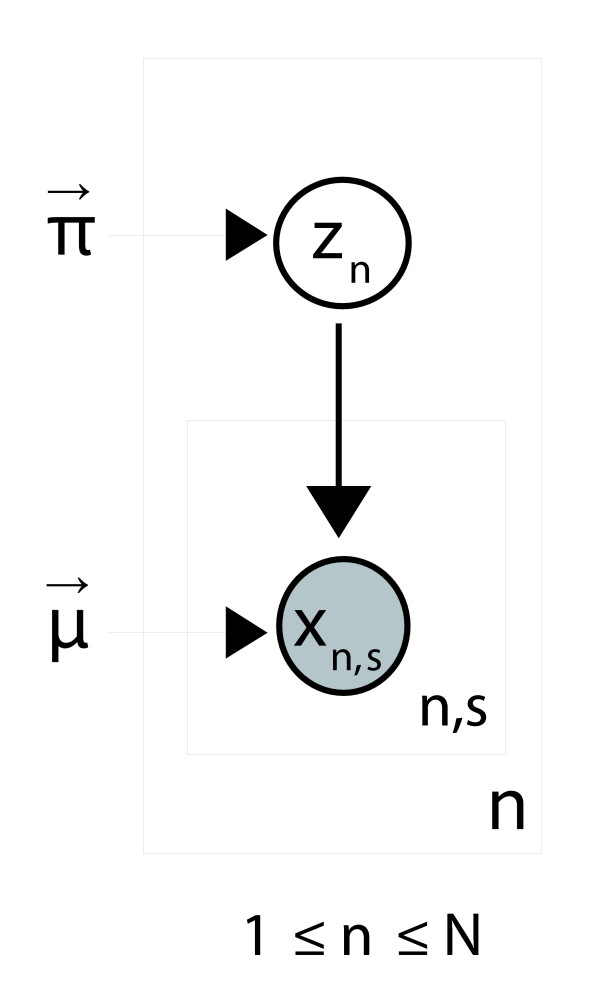
A finite supragenome model was developed to explain the distribution of genes among strains. The model predicts that the NTHi supragenome contains between 4,425 and 6,052 genes with most uncertainty regarding the number of rare genes, those that have a frequency of <0.1 among strains; collectively, these results support the DGH.
分布式基因组假说(DGH)认为,慢性细菌病原体利用多克隆感染和基因特征的重排,以确保在面对适应性宿主防御时能够持续存在。基于多个菌株文库随机测序的研究表明,自由生活的细菌物种拥有一个比任何单个细菌基因组大得多的超基因组。
我们获得了9株不可分型流感嗜血杆菌(NTHi)临床分离株的高深度基因组覆盖,使已测序的NTHi基因组数量达到13个。聚类分析确定了2786个基因,其中1461个基因在所有菌株中都有,其余1328个基因分别存在于部分菌株中;每个菌株的聚类数量在1686至1878个之间。每对菌株之间的基因差异在96至585个之间。将每个NTHi菌株与Rd菌株进行比较,发现每个基因组中有107至158个插入和100至213个缺失。插入和缺失的平均大小分别为1356和1020个碱基对,平均最大插入和缺失分别为26977和37299个碱基对。菌株之间相对大量的小重排与这种天然感受态病原体的转化机制相符。
建立了一个有限超基因组模型来解释基因在菌株间的分布。该模型预测,NTHi超基因组包含4425至6052个基因,其中关于稀有基因(在菌株中频率<0.1的基因)数量的不确定性最大;总体而言,这些结果支持分布式基因组假说。