Lawson-Yuen Amy, Liu Daniel, Han Liqun, Jiang Zhichun I, Tsai Guochuan E, Basu Alo C, Picker Jonathan, Feng Jiamin, Coyle Joseph T
Department of Psychiatry, Harvard Medical School, McLean Hospital, Belmont, MA 02478, USA.
Brain Res. 2007 Nov 14;1180:1-6. doi: 10.1016/j.brainres.2007.08.039. Epub 2007 Aug 24.
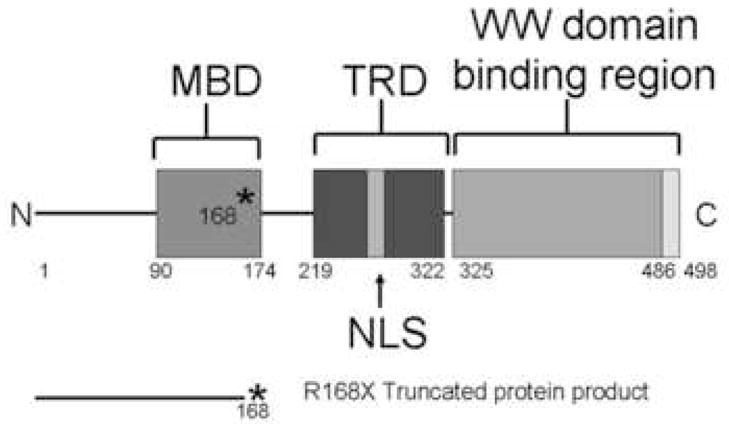
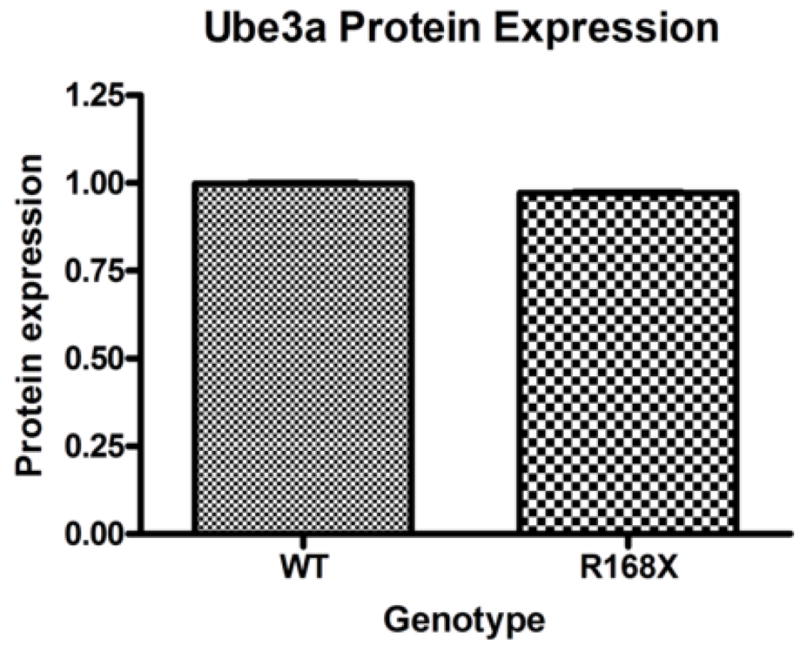
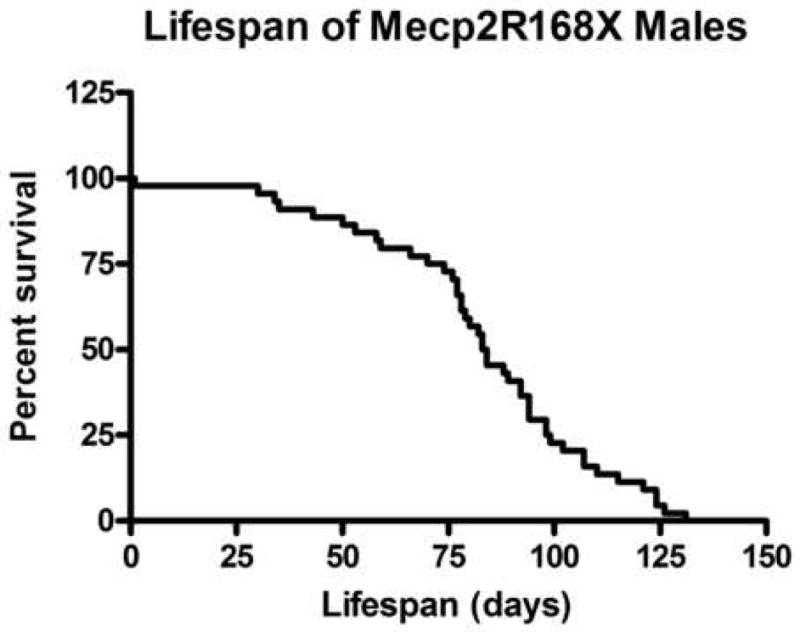
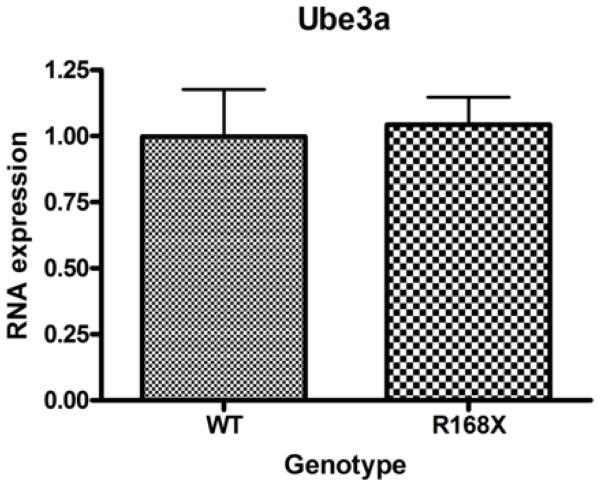
Mutations in the transcriptional repressor methyl CpG binding protein 2 (MeCP2) are responsible for most cases of Rett Syndrome (RS), a severe neurodevelopmental disorder characterized by developmental regression, minimal speech, seizures, postnatal microcephaly and hand stereotypies. Absence of the maternal copy of ubiquitin protein ligase 3A (UBE3A) results in Angelman syndrome, also a severe developmental disorder that shares some clinical features with RS. As MeCP2 regulates gene expression, this has led to the hypothesis that MeCP2 may regulate UBE3A expression; however, there are conflicting reports regarding the expression of Ube3a in MeCP2 null mutant mice. We have generated a novel MeCP2 mutant knock-in mouse with the mutation R168X, one of the most common mutations in patients with RS. These mice show features similar to RS, including hypoactivity, forelimb stereotypies, breathing irregularities, weight changes, hind limb atrophy, and scoliosis. The male mice experience early death. Analysis of Ube3a mRNA and protein levels in the Mecp2(R168X) male mice showed no significant difference in expression compared to their wild type littermates.
转录抑制因子甲基化CpG结合蛋白2(MeCP2)的突变是大多数雷特综合征(RS)病例的病因,RS是一种严重的神经发育障碍,其特征为发育倒退、极少言语、癫痫发作、出生后小头畸形和手部刻板动作。泛素蛋白连接酶3A(UBE3A)母源拷贝的缺失会导致天使综合征,这也是一种严重的发育障碍,与RS有一些共同的临床特征。由于MeCP2调节基因表达,这引发了一种假说,即MeCP2可能调节UBE3A的表达;然而,关于Ube3a在MeCP2基因敲除突变小鼠中的表达存在相互矛盾的报道。我们构建了一种携带R168X突变的新型MeCP2突变基因敲入小鼠,R168X是RS患者中最常见的突变之一。这些小鼠表现出与RS相似的特征,包括活动减少、前肢刻板动作、呼吸不规则、体重变化、后肢萎缩和脊柱侧凸。雄性小鼠过早死亡。对Mecp2(R168X)雄性小鼠中Ube3a mRNA和蛋白水平的分析表明,与野生型同窝小鼠相比,其表达没有显著差异。