Sanders William S, Bridges Susan M, McCarthy Fiona M, Nanduri Bindu, Burgess Shane C
Department of Biochemistry & Molecular Biology, Mississippi State University, MS, USA.
BMC Bioinformatics. 2007 Nov 1;8 Suppl 7(Suppl 7):S23. doi: 10.1186/1471-2105-8-S7-S23.
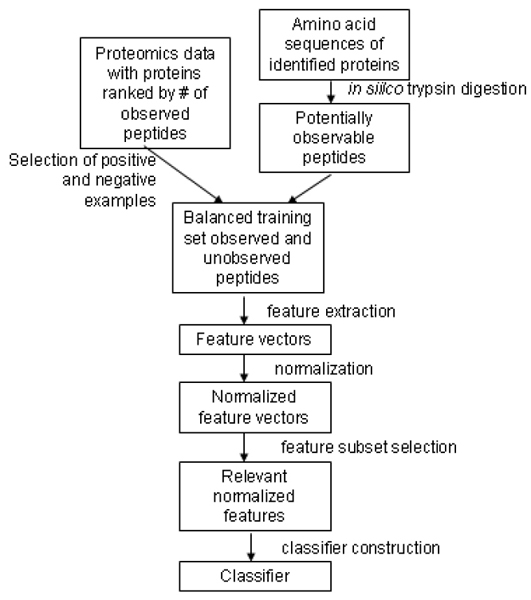
When proteins are subjected to proteolytic digestion and analyzed by mass spectrometry using a method such as 2D LC MS/MS, only a portion of the proteotypic peptides associated with each protein will be observed. The ability to predict which peptides can and cannot potentially be observed for a particular experimental dataset has several important applications in proteomics research including calculation of peptide coverage in terms of potentially detectable peptides, systems biology analysis of data sets, and protein quantification.
We have developed a methodology for constructing artificial neural networks that can be used to predict which peptides are potentially observable for a given set of experimental, instrumental, and analytical conditions for 2D LC MS/MS (a.k.a Multidimensional Protein Identification Technology [MudPIT]) datasets. Neural network classifiers constructed using this procedure for two MudPIT datasets exhibit 10-fold cross validation accuracy of about 80%. We show that a classifier constructed for one dataset has poor predictive performance with the other dataset, thus demonstrating the need for dataset specific classifiers. Classification results with each dataset are used to compute informative percent amino acid coverage statistics for each protein in terms of the predicted detectable peptides in addition to the percent coverage of the complete sequence. We also demonstrate the utility of predicted peptide observability for systems analysis to help determine if proteins that were expected but not observed generate sufficient peptides for detection.
Classifiers that accurately predict the likelihood of detecting proteotypic peptides by mass spectrometry provide proteomics researchers with powerful new approaches for data analysis. We demonstrate that the procedure we have developed for building a classifier based on an individual experimental data set results in classifiers with accuracy comparable to those reported in the literature based on large training sets collected from multiple experiments. Our approach allows the researcher to construct a classifier that is specific for the experimental, instrument, and analytical conditions of a single experiment and amenable to local, condition-specific, implementation. The resulting classifiers have application in a number of areas such as determination of peptide coverage for protein identification, pathway analysis, and protein quantification.
当蛋白质进行蛋白酶解消化并使用二维液相色谱串联质谱法(2D LC MS/MS)等方法进行质谱分析时,与每种蛋白质相关的蛋白型肽段中只有一部分会被观察到。预测特定实验数据集可能观察到和无法观察到哪些肽段的能力在蛋白质组学研究中有几个重要应用,包括根据潜在可检测肽段计算肽段覆盖率、数据集的系统生物学分析以及蛋白质定量。
我们开发了一种构建人工神经网络的方法,可用于预测在二维液相色谱串联质谱法(又称多维蛋白质鉴定技术 [MudPIT])数据集的给定实验、仪器和分析条件下哪些肽段可能被观察到。使用该程序为两个 MudPIT 数据集构建的神经网络分类器在 10 倍交叉验证中的准确率约为 80%。我们表明,为一个数据集构建的分类器对另一个数据集的预测性能较差,从而证明了需要特定于数据集的分类器。除了完整序列的覆盖率百分比外,每个数据集的分类结果还用于根据预测的可检测肽段计算每种蛋白质的信息性氨基酸覆盖率统计数据。我们还证明了预测肽段可观察性在系统分析中的实用性,以帮助确定预期但未观察到的蛋白质是否产生足够的肽段用于检测。
准确预测通过质谱检测蛋白型肽段可能性的分类器为蛋白质组学研究人员提供了强大的新数据分析方法。我们证明,我们基于单个实验数据集构建分类器的程序所得到的分类器,其准确率与基于从多个实验收集的大型训练集的文献报道相当。我们的方法允许研究人员构建一个特定于单个实验的实验、仪器和分析条件的分类器,并且适合于本地、特定条件的实施。所得分类器在许多领域都有应用,如用于蛋白质鉴定的肽段覆盖率测定、通路分析和蛋白质定量。