Rungsarityotin Wasinee, Krause Roland, Schödl Arno, Schliep Alexander
Max Planck Institute for Molecular Genetics, Department of Computational Molecular Biology, Ihnestr, 73, D-14195 Berlin, Germany.
BMC Bioinformatics. 2007 Dec 19;8:482. doi: 10.1186/1471-2105-8-482.
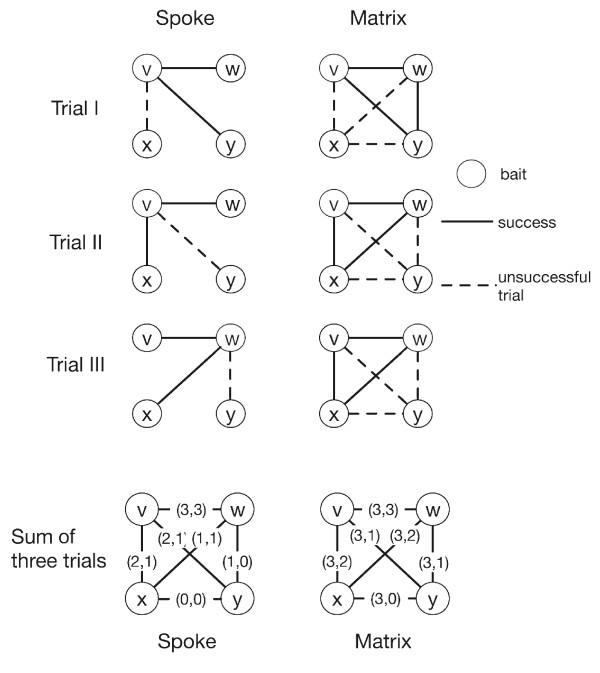
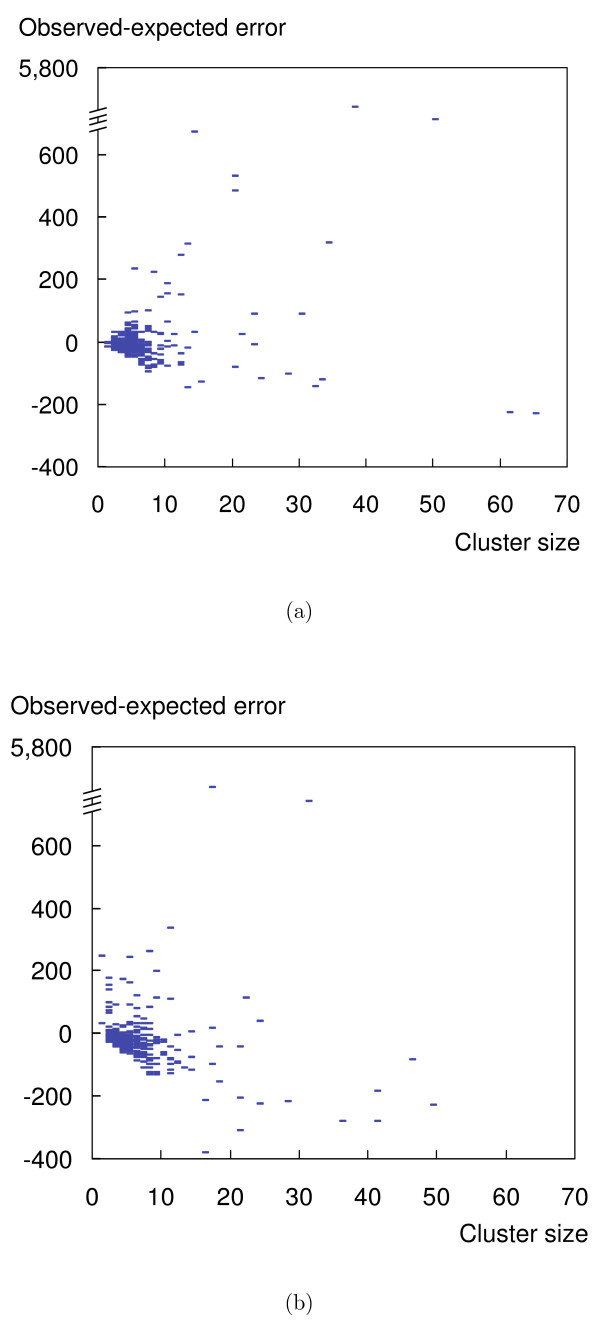
Predicting protein complexes from experimental data remains a challenge due to limited resolution and stochastic errors of high-throughput methods. Current algorithms to reconstruct the complexes typically rely on a two-step process. First, they construct an interaction graph from the data, predominantly using heuristics, and subsequently cluster its vertices to identify protein complexes.
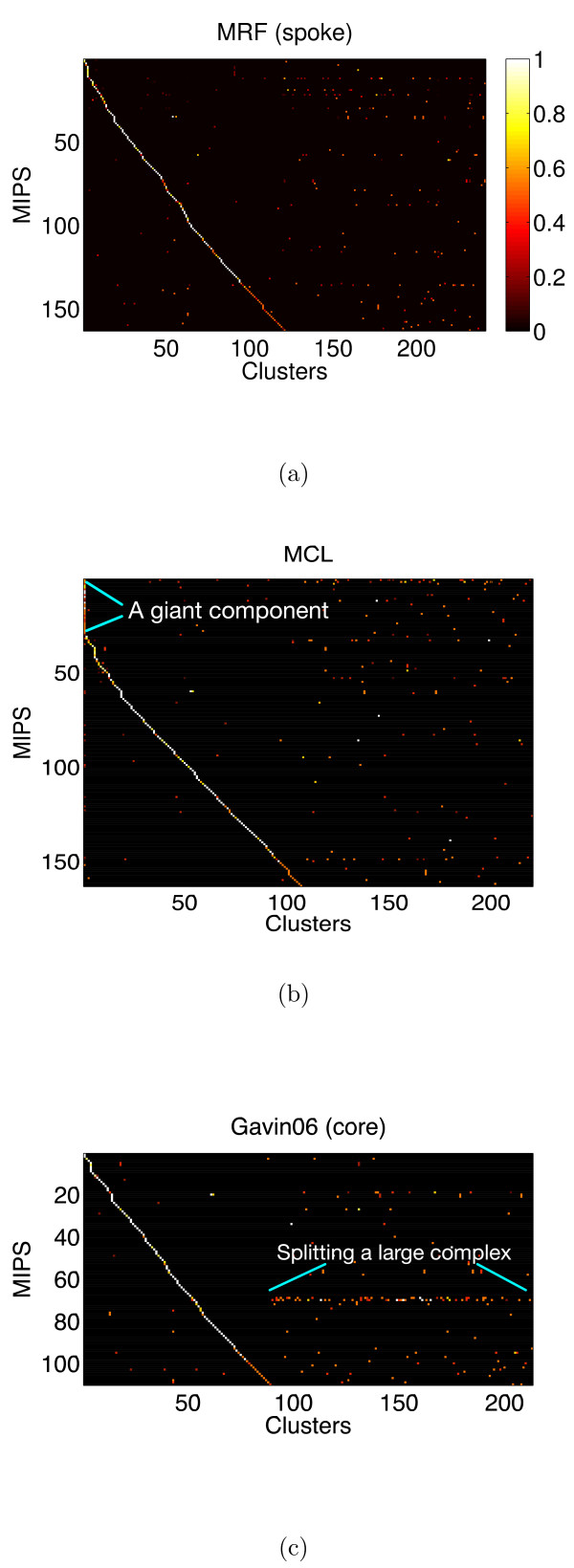
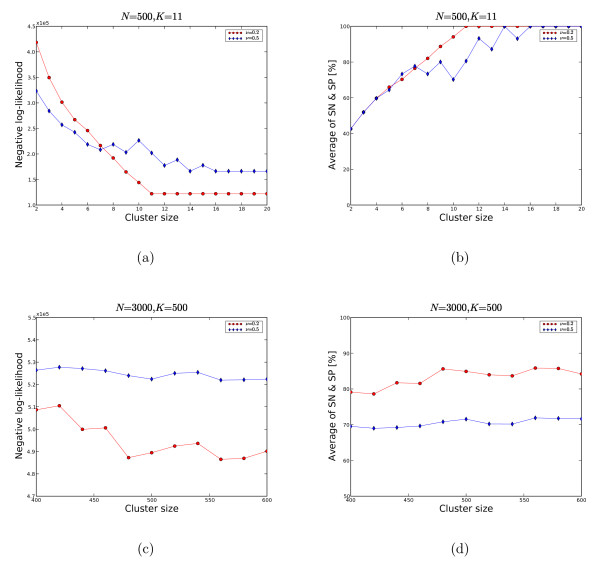
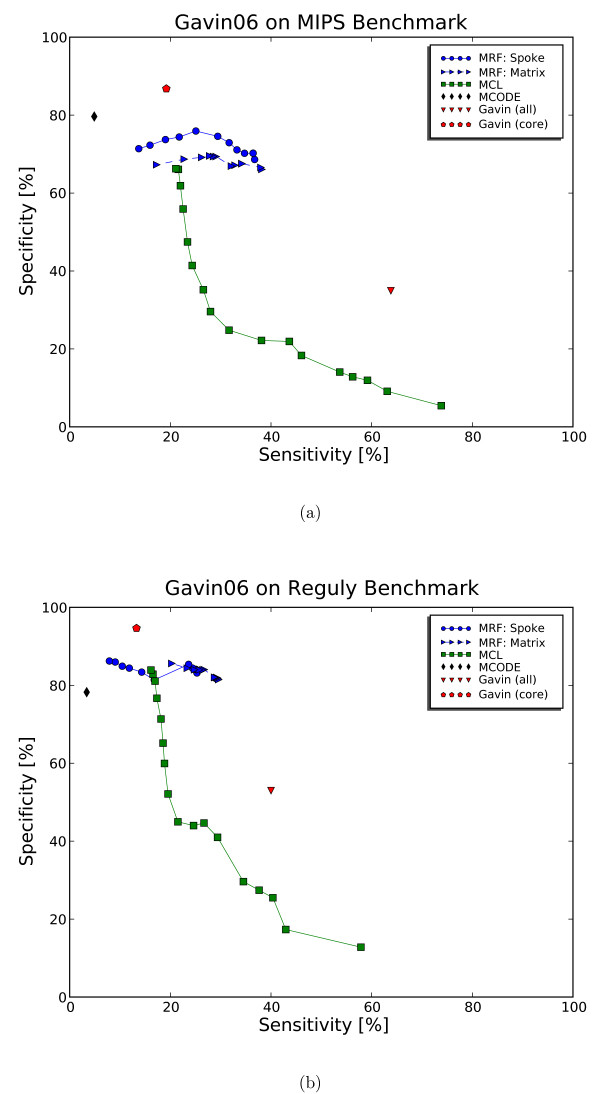
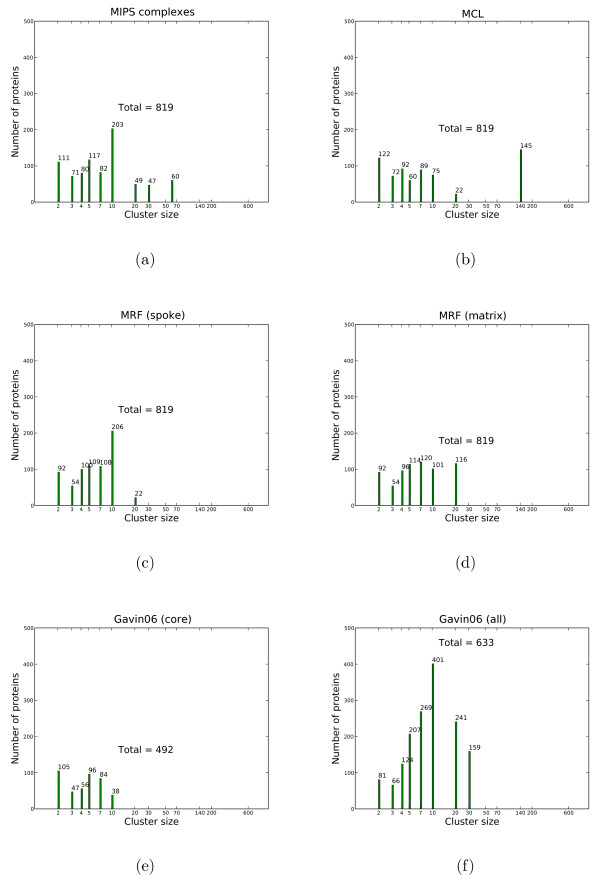
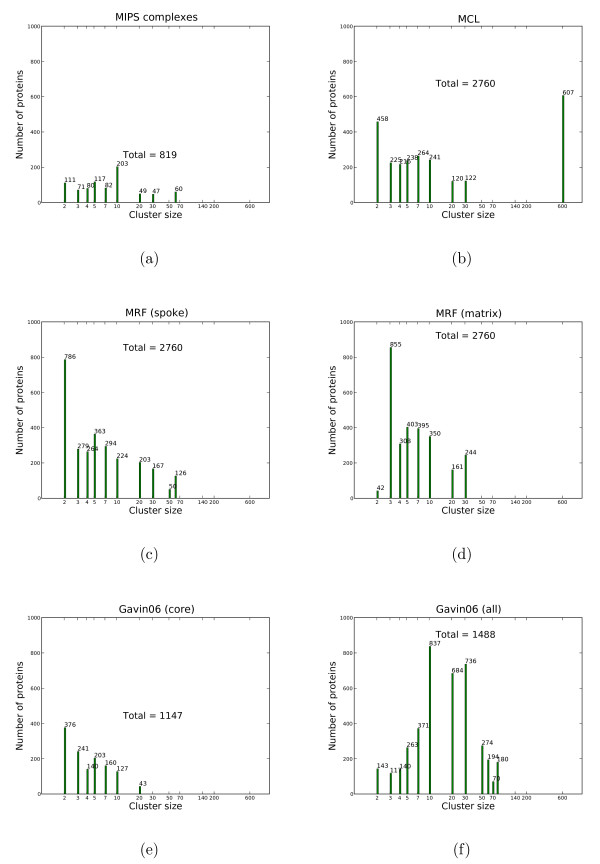
We propose a model-based identification of protein complexes directly from the experimental observations. Our model of protein complexes based on Markov random fields explicitly incorporates false negative and false positive errors and exhibits a high robustness to noise. A model-based quality score for the resulting clusters allows us to identify reliable predictions in the complete data set. Comparisons with prior work on reference data sets shows favorable results, particularly for larger unfiltered data sets. Additional information on predictions, including the source code under the GNU Public License can be found at http://algorithmics.molgen.mpg.de/Static/Supplements/ProteinComplexes.
We can identify complexes in the data obtained from high-throughput experiments without prior elimination of proteins or weak interactions. The few parameters of our model, which does not rely on heuristics, can be estimated using maximum likelihood without a reference data set. This is particularly important for protein complex studies in organisms that do not have an established reference frame of known protein complexes.
由于高通量方法的分辨率有限和随机误差,从实验数据预测蛋白质复合物仍然是一项挑战。当前用于重建复合物的算法通常依赖于两步过程。首先,它们主要使用启发式方法从数据构建相互作用图,随后对其顶点进行聚类以识别蛋白质复合物。
我们提出了一种直接从实验观察中基于模型识别蛋白质复合物的方法。我们基于马尔可夫随机场的蛋白质复合物模型明确纳入了假阴性和假阳性误差,并且对噪声具有高度鲁棒性。为所得聚类基于模型的质量评分使我们能够在完整数据集中识别可靠的预测。与参考数据集上先前工作的比较显示了良好的结果,特别是对于较大的未过滤数据集。有关预测的其他信息,包括遵循GNU公共许可证的源代码,可在http://algorithmics.molgen.mpg.de/Static/Supplements/ProteinComplexes上找到。
我们可以在无需事先去除蛋白质或弱相互作用的情况下,从高通量实验获得的数据中识别复合物。我们的模型不依赖启发式方法,其少数参数可使用最大似然估计,无需参考数据集。这对于没有已知蛋白质复合物既定参考框架的生物体中的蛋白质复合物研究尤为重要。