Flicek Paul
European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB101SD, UK.
Genome Biol. 2007;8(12):233. doi: 10.1186/gb-2007-8-12-233.
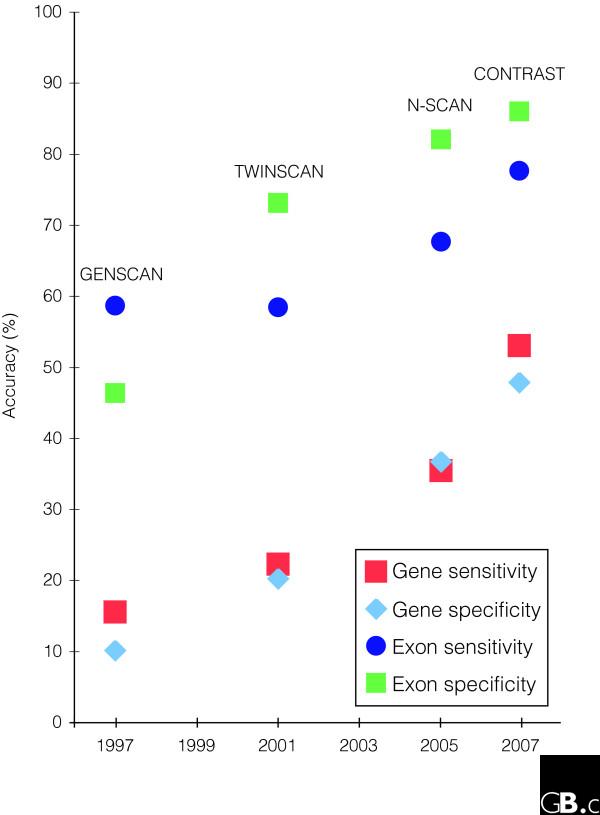
CONTRAST, a new gene-prediction algorithm that uses sophisticated machine-learning techniques, has pushed de novo prediction accuracy to new heights, and has significantly closed the gap between de novo and evidence-based methods for human genome annotation.
CONTRAST是一种使用复杂机器学习技术的新基因预测算法,它将从头预测的准确性提升到了新的高度,并显著缩小了人类基因组注释中从头预测方法与基于证据的方法之间的差距。