Li Jing, Chen Yixuan
Electrical Engineering and Computer Science Department, Case Western Reserve University, Cleveland, OH 44106, USA.
BMC Bioinformatics. 2008 Jan 24;9:44. doi: 10.1186/1471-2105-9-44.
With the completion of the HapMap project, a variety of computational algorithms and tools have been proposed for haplotype inference, tag SNP selection and genome-wide association studies. Simulated data are commonly used in evaluating these new developed approaches. In addition to simulations based on population models, empirical data generated by perturbing real data, has also been used because it may inherit specific properties from real data. However, there is no tool that is publicly available to generate large scale simulated variation data by taking into account knowledge from the HapMap project.
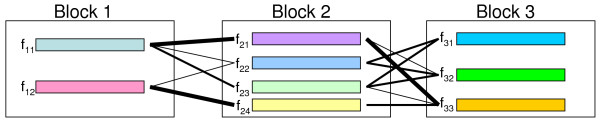
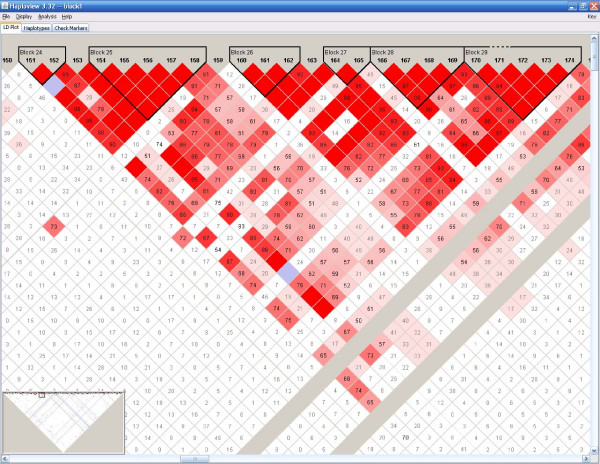
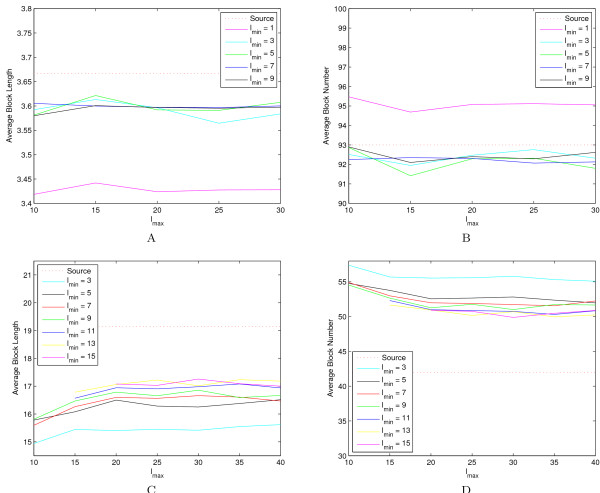
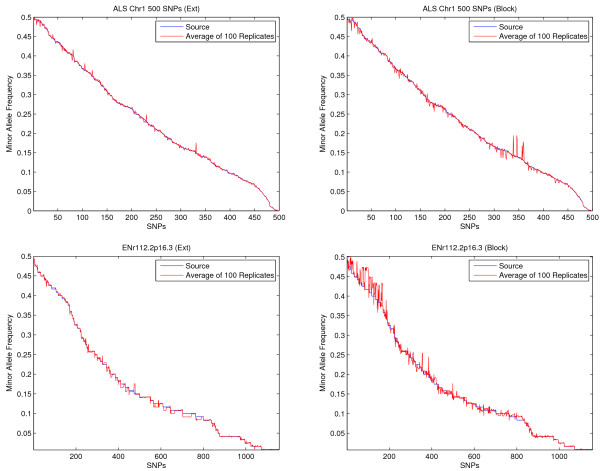
A computer program (gs) was developed to quickly generate a large number of samples based on real data that are useful for a variety of purposes, including evaluating methods for haplotype inference, tag SNP selection and association studies. Two approaches have been implemented to generate dense SNP haplotype/genotype data that share similar local linkage disequilibrium (LD) patterns as those in human populations. The first approach takes haplotype pairs from samples as inputs, and the second approach takes patterns of haplotype block structures as inputs. Both quantitative and qualitative traits have been incorporated in the program. Phenotypes are generated based on a disease model, or based on the effect of a quantitative trait nucleotide, both of which can be specified by users. In addition to single-locus disease models, two-locus disease models have also been implemented that can incorporate any degree of epistasis. Users are allowed to specify all nine parameters in a 3 x 3 penetrance table. For several commonly used two-locus disease models, the program can automatically calculate penetrances based on the population prevalence and marginal effects of a disease that users can conveniently specify.
The program gs can effectively generate large scale genetic and phenotypic variation data that can be used for evaluating new developed approaches. It is freely available from the authors' web site at http://www.eecs.case.edu/~jxl175/gs.html.
随着HapMap计划的完成,已经提出了各种用于单倍型推断、标签SNP选择和全基因组关联研究的计算算法和工具。模拟数据常用于评估这些新开发的方法。除了基于群体模型的模拟之外,通过扰动真实数据生成的经验数据也被使用,因为它可能继承真实数据的特定属性。然而,没有公开可用的工具能够考虑到HapMap计划中的知识来生成大规模模拟变异数据。
开发了一个计算机程序(gs),基于真实数据快速生成大量样本,这些样本可用于多种目的,包括评估单倍型推断、标签SNP选择和关联研究的方法。已实施两种方法来生成与人类群体中具有相似局部连锁不平衡(LD)模式的密集SNP单倍型/基因型数据。第一种方法将样本中的单倍型对作为输入,第二种方法将单倍型块结构模式作为输入。该程序中纳入了数量性状和质量性状。表型基于疾病模型或基于数量性状核苷酸的效应生成,两者均可由用户指定。除了单基因座疾病模型外,还实施了双基因座疾病模型,该模型可纳入任何程度的上位性。允许用户在3×3外显率表中指定所有九个参数。对于几种常用的双基因座疾病模型,该程序可以根据用户可以方便指定的疾病群体患病率和边际效应自动计算外显率。
程序gs可以有效地生成大规模遗传和表型变异数据,可用于评估新开发的方法。可从作者的网站http://www.eecs.case.edu/~jxl175/gs.html免费获取该程序。