Brandsma Corry-Anke, Hylkema Machteld N, van der Strate Barry W A, Slebos Dirk-Jan, Luinge Marjan A, Geerlings Marie, Timens Wim, Postma Dirkje S, Kerstjens Huib A M
Department of Pulmonary Diseases, University Medical Center Groningen, University of Groningen, P,O, Box 30,001, 9700 RB, Groningen, The Netherlands.
Respir Res. 2008 Feb 6;9(1):17. doi: 10.1186/1465-9921-9-17.
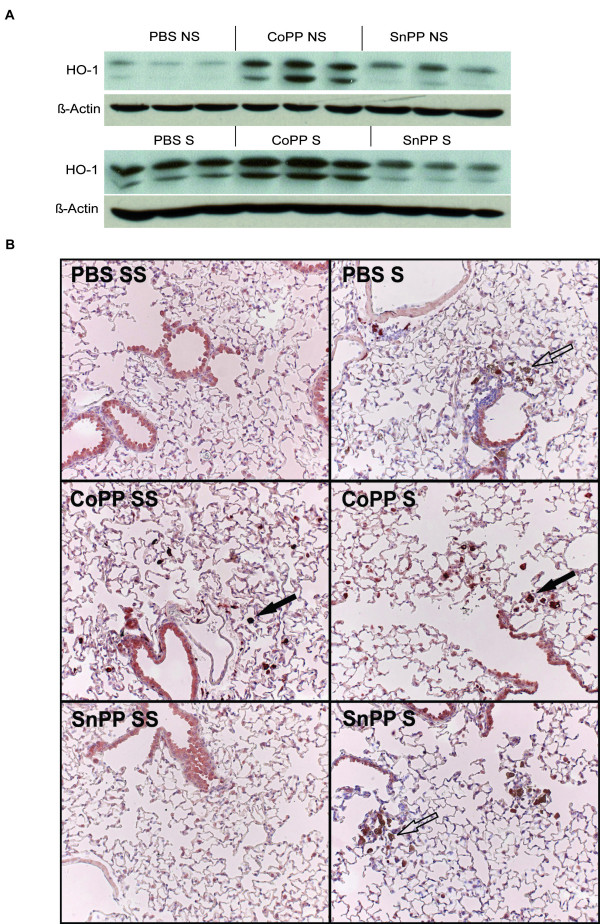
Smoking is the most important cause for the development of COPD. Since not all smokers develop COPD, it is obvious that other factors must be involved in disease development. We hypothesize that heme oxygenase-1 (HO-1), a protective enzyme against oxidative stress and inflammation, is insufficiently upregulated in COPD. The effects of HO-1 modulation on cigarette smoke induced inflammation and emphysema were tested in a smoking mouse model.
Mice were either exposed or sham exposed to cigarette smoke exposure for 20 weeks. Cobalt protoporphyrin or tin protoporphyrin was injected during this period to induce or inhibit HO-1 activity, respectively. Afterwards, emphysema development, levels of inflammatory cells and cytokines, and the presence of B-cell infiltrates in lung tissue were analyzed.
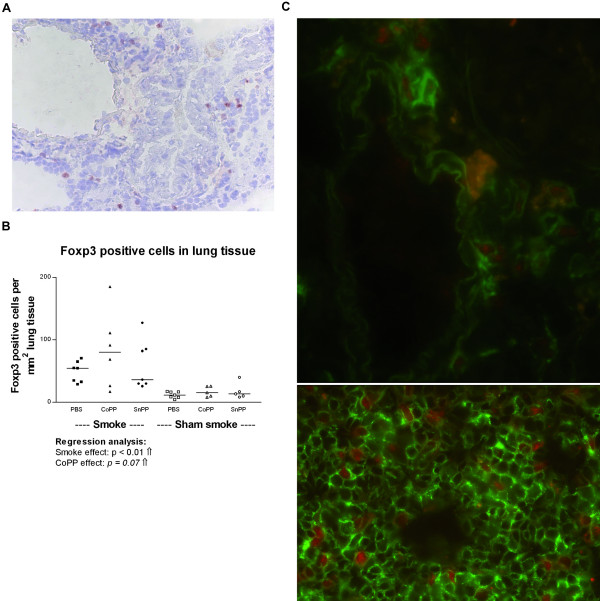
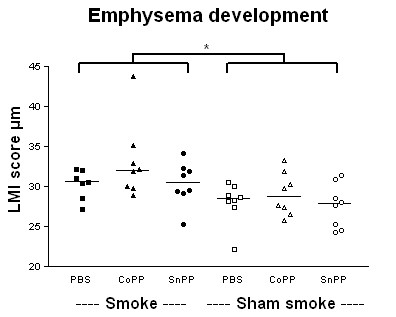
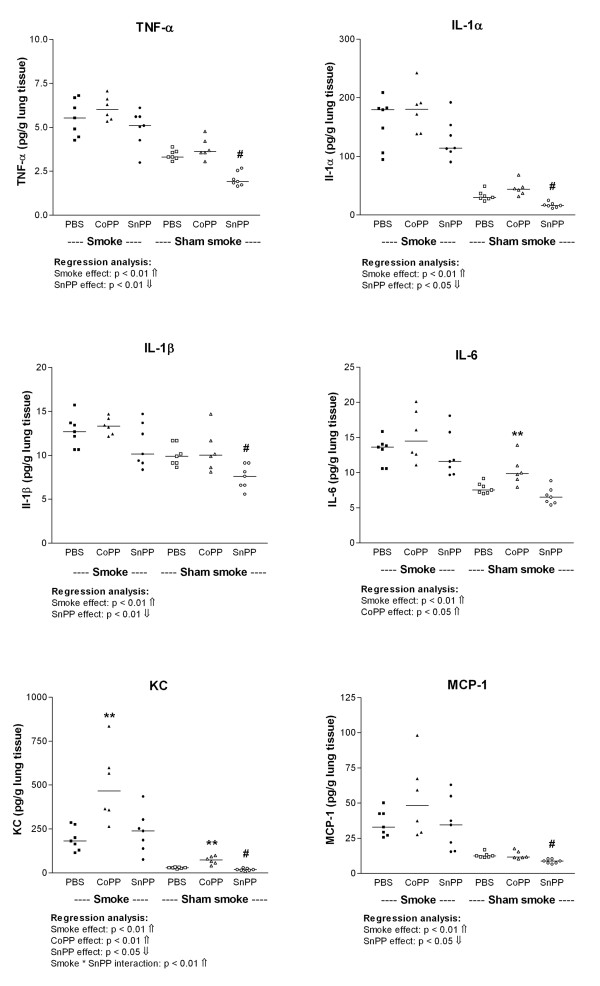
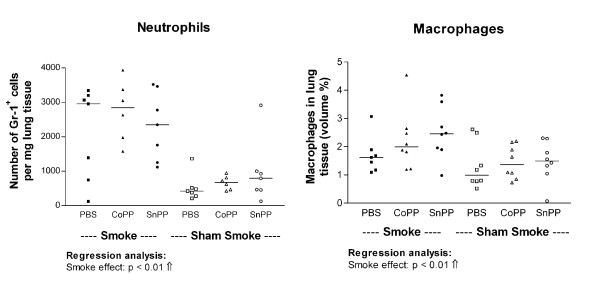
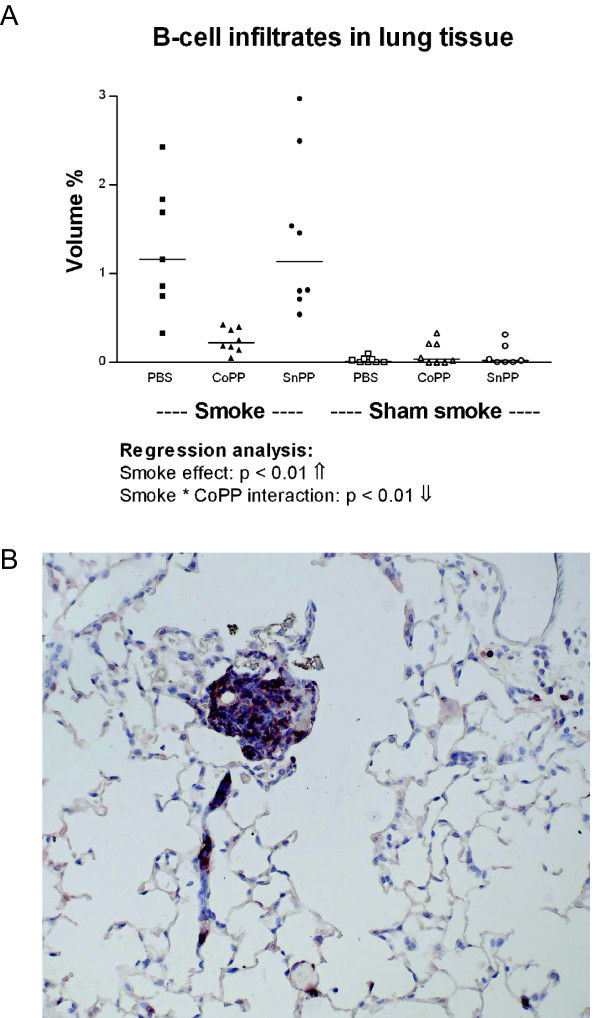
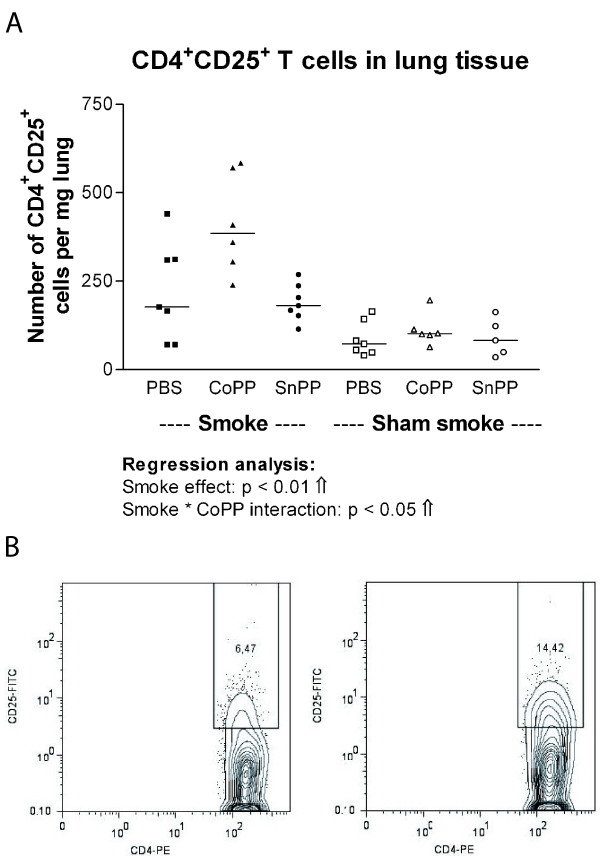
Smoke exposure induced emphysema and increased the numbers of inflammatory cells and numbers of B-cell infiltrates, as well as the levels of inflammatory cytokines in lung tissue. HO-1 modulation had no effects on smoke induced emphysema development, or the increases in neutrophils and macrophages and inflammatory cytokines. Interestingly, HO-1 induction prevented the development of smoke induced B-cell infiltrates and increased the levels of CD4+CD25+ T cells and Foxp3 positive cells in the lungs. Additionally, the CD4+CD25+ T cells correlated positively with the number of Foxp3 positive cells in lung tissue, indicating that these cells were regulatory T cells.
These results support the concept that HO-1 expression influences regulatory T cells and indicates that this mechanism is involved in the suppression of smoke induced B-cell infiltrates. The translation of this interaction to human COPD should now be pursued.
吸烟是慢性阻塞性肺疾病(COPD)发展的最重要原因。由于并非所有吸烟者都会患COPD,显然疾病发展过程中必定涉及其他因素。我们推测,血红素加氧酶-1(HO-1)作为一种抵抗氧化应激和炎症的保护酶,在COPD中上调不足。在吸烟小鼠模型中测试了HO-1调节对香烟烟雾诱导的炎症和肺气肿的影响。
将小鼠暴露于香烟烟雾或假暴露于香烟烟雾20周。在此期间分别注射钴原卟啉或锡原卟啉以诱导或抑制HO-1活性。之后,分析肺气肿的发展、炎症细胞和细胞因子水平以及肺组织中B细胞浸润情况。
烟雾暴露诱导肺气肿,增加炎症细胞数量、B细胞浸润数量以及肺组织中炎症细胞因子水平。HO-1调节对烟雾诱导的肺气肿发展、中性粒细胞和巨噬细胞数量增加以及炎症细胞因子无影响。有趣的是,HO-1诱导可防止烟雾诱导的B细胞浸润发展,并增加肺中CD4 + CD25 + T细胞和Foxp3阳性细胞水平。此外,CD4 + CD25 + T细胞与肺组织中Foxp3阳性细胞数量呈正相关,表明这些细胞是调节性T细胞。
这些结果支持HO-1表达影响调节性T细胞的概念,并表明该机制参与抑制烟雾诱导的B细胞浸润。现在应该探索这种相互作用在人类COPD中的转化情况。